
Author: Lubomir Vezenkov

Titouan received a prize for his innovativeness !

From the IBMM team

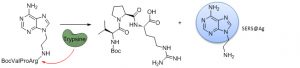
Synthesis of Peptide–Adenine Conjugates as a New Tool for Monitoring Protease Activity
How are 1,2,3-triazoles accommodated in helical secondary structures?
12/10-Helix in Mixed β-Peptides Alternating Bicyclic and Acyclic β-Amino Acids: Probing the Relationship between Bicyclic Side Chain and Helix Stability
Congratulation Nicolas! New paper in Eur. J. Org. Chem.
Merlion Grant : collaboration between peptide team and NTU in tissue engineering

