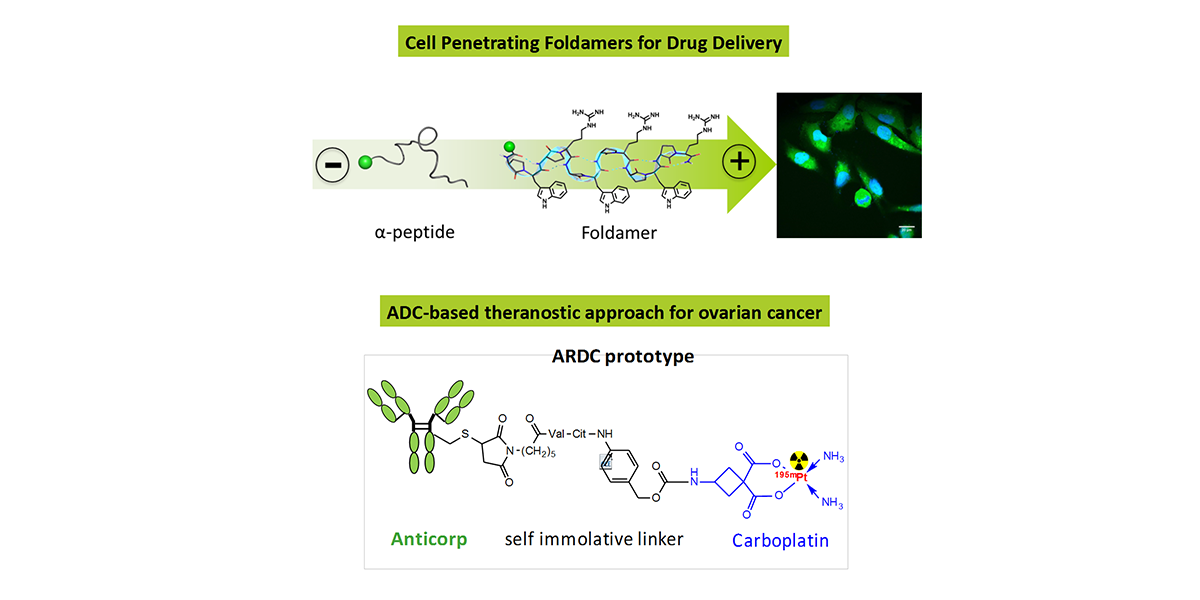
A lot of potential drugs are ineffective because of their inability to cross certain biological membranes, such as the lipid bilayer or the blood brain barrier. Once inside the cell, those compounds often have to find a local address, also known as cell compartment, but unfortunately often they will lose their way and find themselves trapped in the ‘’wrong neighborhoods’’ (cell organelles). In the same time most of the anti-cancer drugs have devastating side effects due to the fact that they target equally healthy and tumour cell. For all those reasons the terms – ‘’Vectorization, targeting and drug delivery’’ are key words in modern medicinal chemistry. In our group we actively pursue the development of cell penetrating and cell targeting compounds based on peptides, foldamers and anti-bodies. These vectors are used to send bioactive compounds, such as anti-cancer drugs, to a preferred cellular compartment or to target a certain organ or cell type in the living species.







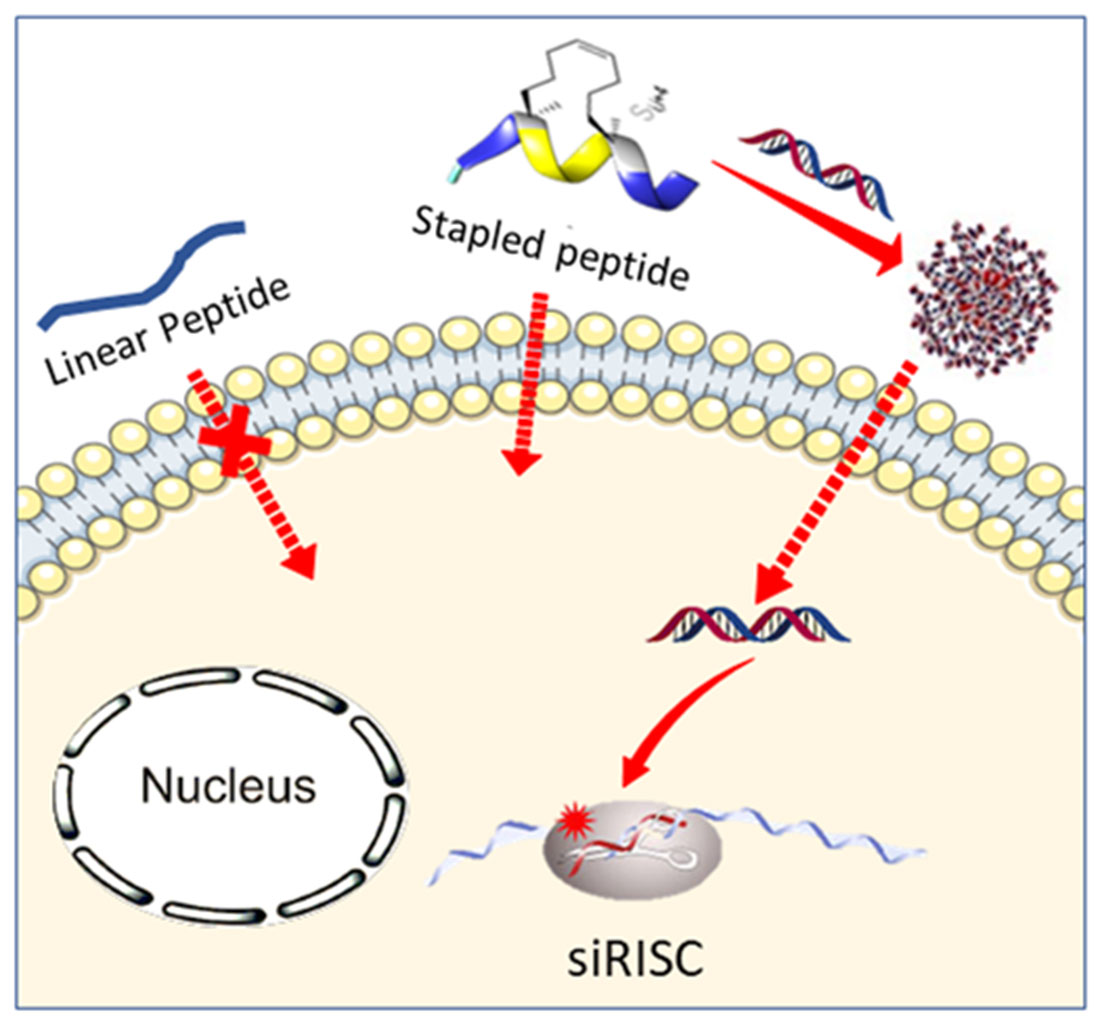
Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery.
Nanomaterials (Basel). 2020 Nov 25;10(12):2334. doi: 10.3390/nano10122334
Simon M, Laroui N, Heyraud M, Laconde G, Ali LMA, Bourbiaux K, Subra G, Vezenkov LL, Legrand B, Amblard M, Bettache N.
Abstract
Small interfering RNAs (siRNAs) are promising molecules for developing new therapies based on gene silencing; however, their delivery into cells remains an issue. In this study, we took advantage of stapled peptide technology that has emerged as a valuable strategy to render natural peptides more structured, resistant to protease degradation and more bioavailable, to develop short carriers for siRNA delivery. From the pool of stapled peptides that we have designed and synthesized, we identified non-toxic vectors that were able to efficiently encapsulate siRNA, transport them into the cell and induce gene silencing. Remarkably, the most efficient stapled peptide (JMV6582), is composed of only eight amino-acids and contains only two cationic charges.

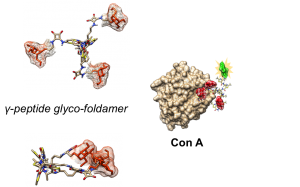
Can Heterocyclic γ-Peptides Provide Polyfunctional Platforms for Synthetic Glycocluster Construction?
Chemistry. 2018 Aug 6;24(44):11426-11432. doi: 10.1002/chem.201802032
Simon M, Ali LMA, El Cheikh K, Aguesseau J, Gary-Bobo M, Garcia M, Morère A, Maillard LT.
Abstract
Sugars play key roles in many molecular and cellular communication processes involving a family of proteins named lectins. The low affinity associated with sugar recognition is generally counterbalanced by the multivalent nature of the interaction. While many polyglycosylated architectures have been described, only a few studies focused on the impact of topology variations of the multivalent structures on the interaction with lectin proteins. One major interest of our group concerns the design of new highly predictable and stable molecular pseudo‐peptide architectures for therapeutic applications. In such a context, we described a class of constrained heterocyclic γ‐amino acids built around a thiazole ring, named ATCs. ATC oligomers are helical molecules resulting from the formation of a highly stable C9 hydrogen‐bonding pattern. Following our program, we herein address the potential of ATC oligomers as tunable scaffolds for the development of original multivalent glycoclusters.
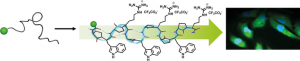
Ribbon-like Foldamers for Cellular Uptake and Drug Delivery
Chembiochem 2017 Nov 2;18(21):2110-2114. doi: 10.1002/cbic.201700455. Epub 2017 Sep 22.
Vezenkov LL, Martin V, Bettache N, Simon M, Messerschmitt A, Legrand B, Bantignies JL, Subra G, Maynadier M, Bellet V, Garcia M, Martinez J, Amblard M.
Abstract
Different intracellular delivery systems of bioactive compounds have been developed, including cell-penetrating peptides. Although usually nontoxic and biocompatible, these vectors share some of the general drawbacks of peptides, notably low bioavailability and susceptibility to protease degradation, that limit their use. Herein, the conversion of short peptide sequences into poly-α-amino-γ-lactam foldamers that adopt a ribbon-like structure is investigated. This template is used to distribute critical cationic and/or hydrophobic groups on both sides of the backbone, leading to potent short, cell-permeable foldamers with a low positive-charge content. The lead compound showed dramatically improved protease resistance and was able to efficiently deliver a biologically relevant cargo inside cells. This study provided a simple strategy to convert short peptide sequences into efficient protease-resistant cell-penetrating foldamers.
Structure-Activity Relationships of JMV4463, a Vectorized Cathepsin D Inhibitor with Antiproliferative Properties: The Unique Role of the AMPA-Based Vector
ChemMedChem, 2016, Volume: 11, Issue: 3, Pages: 302-308, DOI: 10.1002/cmdc.201500457
L. Vezenkov, C. A. Sanchez, V. Bellet, V. Martin, M. Maynadier, N. Bettache, V. Lisowski, J. Martinez, M. Garcia, M. Amblard, J. F. Hernandez
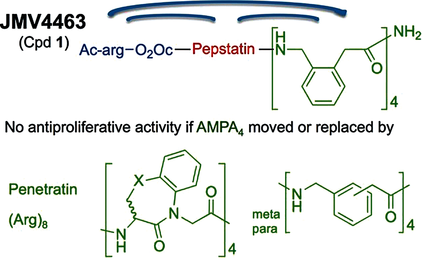
Abstract
Cathepsin D (CathD) is overexpressed and secreted by several solid tumors and stimulates their growth, the mechanism of which is still not understood. In this context, the pepstatin bioconjugate JMV4463 [Ac-arg-O2Oc-(Val)3-Sta-Ala-Sta-(AMPA)4-NH2; O2Oc=8-amino-3,6-dioxaoctanoyl, Sta=statine, AMPA=ortho-aminomethylphenylacetyl], contg. a new kind of cell-penetrating vector, was previously shown to exhibit potent antiproliferative effects in vitro and to delay the onset of tumors in vivo. In this study, the authors performed a structure-activity relationship anal. to evaluate the significance of the inhibitor and vector moieties of JMV4463. By modifying both statine residues of pepstatin the authors found that the antiproliferative activity is correlated with CathD inhibition, supporting a major role of the catalytic activity of intracellular CathD in cancer cell proliferation. Replacing the vector composed of four AMPA units with other vectors was found to abolish cytotoxicity, although all of the conjugates enabled pepstatin transport into cells. In addn., the AMPA4 vector must be localized at the C terminus of the bioconjugate. The unexpected importance of the vector structure and position for cytotoxic action suggests that AMPA4 enables pepstatin to inhibit the proteolysis of crit. CathD substrates involved in cell proliferation via a unique mechanism of action.
Dipeptide mimic oligomer transporter mediates intracellular delivery of Cathepsin D inhibitors: a potential target for cancer therapy
J Control Release. 2013 Oct 28;171(2):251-7. doi: 10.1016/j.jconrel.2013.07.017. Epub 2013 Jul 27.
Maynadier M, Vezenkov LL, Amblard M, Martin V, Gandreuil C, Vaillant O, Gary-Bobo M, Basile I, Hernandez JF, Garcia M, Martinez J.
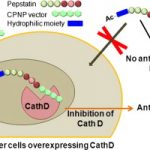
Abstract
Implication of the intracellular proteolytic activity of Cathepsin D (CathD), a lysosomal aspartyl-protease overexpressed in numerous solid tumors, has been evidenced on tumor growth. Its intracellular inhibition by potent inhibitors such as pepstatin constitutes a relevant but challenging molecular target. Indeed the potential of pepstatin as a therapeutic molecule is hampered by its too low intracellular penetration. We addressed this limitation by designing and developing a bioconjugate combining a pepstatin derivative with a new vector of cell penetration (CPNP) specifically targeting the endolysosomal compartment. We showed that this pepstatin conjugate (JMV4463) exhibited high anti-proliferative effect on tumor cell cultures via intracellular CathD inhibition and altered cell cycle associated with apoptotic events in vitro. When tested in mice xenografted with breast cancer cells, JMV4463 delayed tumor emergence and growth.
