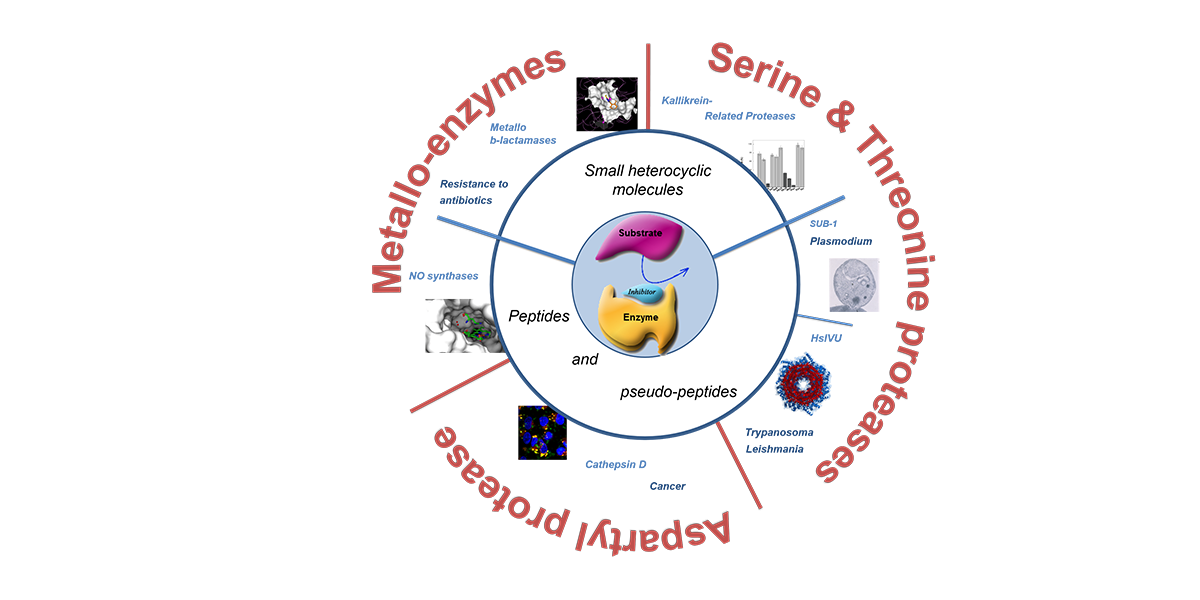
Several projects in medicinal chemistry deal with enzyme inhibition. Enzymes play central roles in all life processes and in some circumstances, their inhibition can help resolve pathological situations. In most diseases, if not all, it is possible to propose at least one enzyme as a potential target, and many drugs are enzyme inhibitors. Enzymes could be human enzymes that can be inhibited to treat various diseases as cardio-vascular, inflammatory, cancer and neurological diseases. Enzymes can also be exogenous ones that are essential for the life of deadly virus, bacteria or parasites.
We have a long time experience in the design of enzyme inhibitors and we are currently investigating the following medicinal targets:
– NO Synthases
– Metallo-β-lactamases (MBL) and the bacterial resistance to β-lactam antibiotics
– HslVU, a proteasome-like complex present in the mitochondria of Trypanosomatids
– Cancers with overexpression of the lysosomal Cathepsin D (CathD)
– Kallikrein inhibitors
– New antimelanoma agents
CONTACT







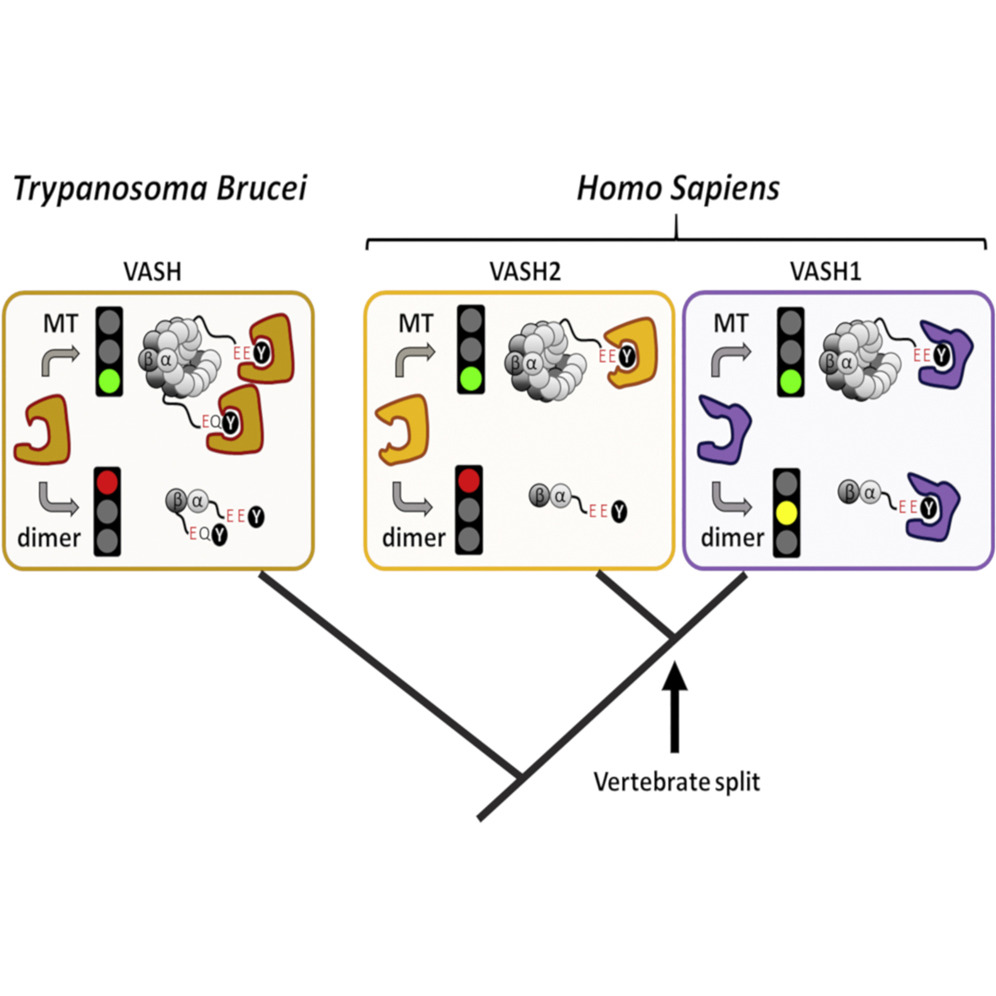
Evolutionary Divergence of Enzymatic Mechanisms for Tubulin Detyrosination
Cell Rep. 2019 Dec 17;29(12):4159-4171.e6. doi: 10.1016/j.celrep.2019.11.074.
Van der Laan S, Lévêque MF, Marcellin G, Vezenkov L, Lannay Y, Dubra G, Bompard G, Ovejero S, Urbach S, Burgess A, Amblard M, Sterkers Y, Bastien P, Rogowski K.
Abstract
The two related members of the vasohibin family, VASH1 and VASH2, encode human tubulin detyrosinases. Here we demonstrate that, in contrast to VASH1, which requires binding of small vasohibin binding protein (SVBP), VASH2 has autonomous tubulin detyrosinating activity. Moreover, we demonstrate that SVBP acts as a bona fide activator of both enzymes. Phylogenetic analysis of the vasohibin family revealed that regulatory diversification of VASH-mediated tubulin detyrosination coincided with early vertebrate evolution. Thus, as a model organism for functional analysis, we used Trypanosoma brucei (Tb), an evolutionarily early-branched eukaryote that possesses a single VASH and encodes a terminal tyrosine on both α- and β-tubulin tails, both subject to removal. Remarkably, although detyrosination levels are high in the flagellum, TbVASH knockout parasites did not present any noticeable flagellar abnormalities. In contrast, we observed reduced proliferation associated with profound morphological and mitotic defects, underscoring the importance of tubulin detyrosination in cell division.
Synthesis of Peptide–Adenine Conjugates as a New Tool for Monitoring Protease Activity
Eur. J. Org. Chem. January 2019: 176-183. doi:10.1002/ejoc.201801490.
Masurier, N. , Soualmia, F. , Sanchez, P. , Lefort, V. , Roué, M. , Maillard, L. T., Subra, G. , Percot, A. and El Amri, C.

Abstract
We took advantage of the powerful adenine SERS (Surface Enhanced Raman Spectroscopy) probe to design peptide–adenine conjugates as candidates for use as serine protease substrates. Whereas the direct introduction of the peptide sequence on the adenine exocyclic N6 amine gave an imidazopurinone derivative, the introduction of an aminoethyl linker between the adenine group and the peptide chain led to the expected candidate probes. These potential substrates were then evaluated for monitoring the hydrolytic activity of trypsin, used as a model protease, by HPLC and by SERS. We demonstrated that the Boc–VPR–adenine conjugate is a substrate of trypsin and constitutes a good starting point to design optimized substrates to monitor protease activity by SERS.
Inhibitors of kallikrein-related peptidases: An overview.
Med Res Rev. 2018 Mar;38(2):655-683. doi: 10.1002/med.21451.
Masurier N, Arama DP, El Amri C, Lisowski V.
Abstract
Kallikrein-related peptidases (KLKs) are a family of 15 secreted serine proteases that are involved in various physiological processes. Their activities are subtly regulated by various endogenous inhibitors, ranging from metallic ions to macromolecular entities such as proteins. Furthermore, dysregulation of KLK activity has been linked to several pathologies, including cancer and skin and inflammatory diseases, explaining the numerous efforts to develop KLK-specific pharmacological inhibitors as potential therapeutic agents. In this review, we focus on the huge repertoire of KLKs inhibitors reported to date with a special emphasis on the diversity of their molecular mechanisms of inhibition.
1,2,4-Triazole-3-thione Compounds as Inhibitors of Dizinc Metallo-β-lactamases
ChemMedChem. 2017 Jun 21;12(12):972-985. doi: 10.1002/cmdc.201700186. Epub 2017 Jun 12.
Sevaille L, Gavara L, Bebrone C, De Luca F, Nauton L, Achard M, Mercuri P, Tanfoni S, Borgianni L, Guyon C, Lonjon P, Turan-Zitouni G, Dzieciolowski J, Becker K, Bénard L, Condon C, Maillard L, Martinez J, Frère JM, Dideberg O, Galleni M, Docquier JD, Hernandez JF.
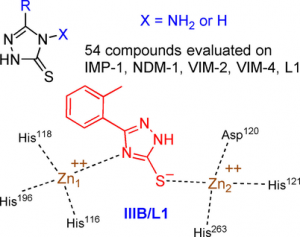
Abstract
Metallo-β-lactamases (MBLs) cause resistance of Gram-negative bacteria to β-lactam antibiotics and are of serious concern, because they can inactivate the last-resort carbapenems and because MBL inhibitors of clinical value are still lacking. We previously identified the original binding mode of 4-amino-2,4-dihydro-5-(2-methylphenyl)-3H-1,2,4-triazole-3-thione (compound IIIA) within the dizinc active site of the L1 MBL. Herein we present the crystallographic structure of a complex of L1 with the corresponding non-amino compound IIIB (1,2-dihydro-5-(2-methylphenyl)-3H-1,2,4-triazole-3-thione). Unexpectedly, the binding mode of IIIB was similar but reverse to that of IIIA. The 3 D structures suggested that the triazole-thione scaffold was suitable to bind to the catalytic site of dizinc metalloenzymes. On the basis of these results, we synthesized 54 analogues of IIIA or IIIB. Nineteen showed IC50 values in the micromolar range toward at least one of five representative MBLs (i.e., L1, VIM-4, VIM-2, NDM-1, and IMP-1). Five of these exhibited a significant inhibition of at least four enzymes, including NDM-1, VIM-2, and IMP-1. Active compounds mainly featured either halogen or bulky bicyclic aryl substituents. Finally, some compounds were also tested on several microbial dinuclear zinc-dependent hydrolases belonging to the MBL-fold superfamily (i.e., endonucleases and glyoxalase II) to explore their activity toward structurally similar but functionally distinct enzymes. Whereas the bacterial tRNases were not inhibited, the best IC50 values toward plasmodial glyoxalase II were in the 10 μm range.
Conformationally Constrained Peptidomimetics as Inhibitors of the Protein Arginine Methyl Transferases
Chemistry – A European Journal, 2016, Volume: 22, Issue: 39, Pages: 14022-14028, DOI: 10.1002/chem.201602518
A. Knuhtsen, B. Legrand, O. Van der Poorten, M. Amblard, J. Martinez, S. Ballet, J. L. Kristensen, D. S. Pedersen
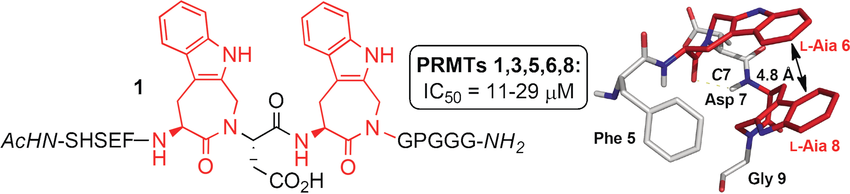
Abstract
Protein arginine N-Me transferases (PRMTs) belong to a family of enzymes that modulate the epigenetic code through modifications of histones. In the present study, peptides emerging from a phage display screening were modified in the search for PRMT inhibitors through substitution with non-proteinogenic amino acids, N-alkylation of the peptide backbone, and incorporation of constrained dipeptide mimics. One of the modified peptides (23) showed an increased inhibitory activity towards several PRMTs in the low μM range and the conformational preference of this peptide was investigated and compared with the original hit using CD and NMR spectroscopy. Introducing two constrained tryptophan residue mimics (L-Aia) spaced by a single amino acid was found to induce a unique turn structure stabilized by a hydrogen bond and arom. π-stacking interaction between the two L-Aia residues.
Pyrido-imidazodiazepinones as a new class of reversible inhibitors of human kallikrein 7
European Journal of Medicinal Chemistry, 2015, Volume: 93, Pages: 202-213, DOI: 10.1016/j.ejmech.2015.02.008
D. P. Arama, F. Soualmia, V. Lisowski, J.-F. Longevial, E. Bosc, L. T. Maillard, J. Martinez, N. Masurier, C. El Amri
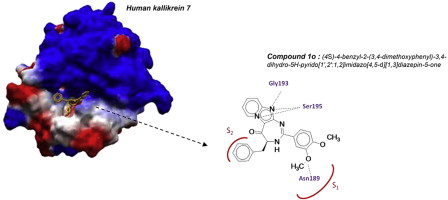
Abstract
The human tissue kallikrein-7 (KLK7) is a chymotryptic serine protease member of tissue kallikrein family. KLK7 is involved in skin homeostasis and inflammation. Excess of KLK7 activity is also assocd. with tumor metastasis processes, esp. in ovarian carcinomas, prostatic and pancreatic cancers. Development of Kallikrein 7 inhibitors is thus of great interest in oncol. but also for treating skin diseases. Most of the developed synthetic inhibitors present several drawbacks such as poor selectivity and unsuitable physico-chem. properties for in vivo use. Recently, the authors described a practical sequence for the synthesis of imidazopyridine-fused [1,3]-diazepines. Here, the authors report the identification of pyrido-imidazodiazepinone core as a new potential scaffold to develop selective and competitive inhibitors of kallikrein-related peptidase 7. Structure-activity relationships (SAR), inhibition mechanisms and selectivity as well as cytotoxicity against selected cancer cell lines were investigated.
Imidazopyridine-fused [1,3]-diazepinones: Synthesis and antiproliferative activity
European Journal of Medicinal Chemistry, 2014, Volume: 75, Pages: 382-390, DOI: 10.1016/j.ejmech.2014.01.044
A. Gallud, O. Vaillant, L.T. Maillard, D. Arama, J. Dubois, M. Maynadier, V. Lisowski, M. Garcia, J. Martinez, N. Masurier
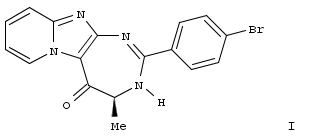
Abstract
A series of 15 pyrido-imidazo-1,3-diazepin-5-ones and pyrido-1,3-diazepine-2,5-diones were synthesized and their anticancer activities were evaluated. Among tested compds. on a cell lines panel, compd. I presents the best growth inhibition activity on 21 cell lines with a cytotoxic effect on MDA-MB-435 melanoma cells. This compd. led to deep cell morphol. changes and revealed to be an inhibitor of the Hepatocyte progenitor kinase-like kinase (HGK), which is known to be implicated in the migration, adhesion and invasion of various tumor cells.
