
MURIEL AMBLARD
Senior researcher, CNRS MONTPELLIER
Dr Muriel AMBLARD obtained her PhD in Organic Chemistry at the University of Montpellier under the supervision of Prof Jean Martinez in 1991. She joined the group of Dr Paul Anderson at Merck Sharp & Dohm in West point (Pennsylvania, USA) as a post-doctoral fellow. In 1996, she obtained a position at the Laboratory of Amino Acid Peptide and Protein in Montpellier as a CNRS researcher. She became a CNRS senior researcher and a team leader at the Institute of Biomolecules Max Mousseron in 2007. Her research interests are mainly at the interface of chemistry, biology and analysis. She developed a number of potent receptor agonists and antagonists of several peptide hormones. Related to medicinal chemistry programs, she developed new multifunctional scaffolds and focused on the design and synthesis of small molecules (diazepinones, spiroimidazolidinones, constraint dipeptide mimics) by combinatorial chemistry on solid support.
Her recent work focuses on the design and synthesis of constrained β-amino acid and dipeptide mimic oligomers as highly predictable and stable helical molecular architectures (Foldamers) applied to 1) the identification of cell penetrating compounds for the delivery of bioactive compounds inside the cells and 2) inhibitors of protein/protein interactions.
Another part of her research is devoted to the development of (1) new methodologies in peptide synthesis (the use of an O-N acyl transfer reaction for the synthesis of cyclic peptides without epimerization, new strategy for the synthesis of peptides alcohols on solid support, synthesis of stapled peptides ans peptide-polymers….) and (2) peptide-based biopolymers by new approaches relying on bottom up polymerisation of peptide-activated monomer.
Muriel is co-author of over 85 papers, 5 book chapters and 7 patents.
Contact:
muriel.amblard@umontpellier.fr
0033411759605
5 major publications :
D. Paramelle, G. Subra, L. Vezenkov, M. Maynadier, C. André, C. Enjalbal, M. Calmès, M. Garcia, J. Martinez, M. Amblard. A straightforward approach for cellular uptake quantification. Angew. Chem. Int. Ed. 2010, 49, 8240-8243.
B. Legrand, C. André, E. Wenger, C. Didierjean, M C. Averlant-Petit, J. Martinez, M. Calmes, M. Amblard.Robust Helix Formation in a New Family of oligoureas based on a Constrained Bicyclic Building Block, Angew. Chem. Int. Ed., 2012, 51, 11267-11270
B. Legrand, C. André, L. Moulat, E. Wenger, C. Didierjean, E. Aubert, M C. Averlant-Petit, J. Martinez, M. Calmes, M. Amblard.Unprecedented Chain-Length Dependent Conformational Conversion Between 11/9 and 18/16 Helix inα/β-Hybrid Peptides. Angew. Chem. Int. Ed., 2014, 53, 13131-13135.
A. Martin, B. Legrand, L. L. Vezenkov, M. Berthet, G. Subra, M. Calmès, J.L. Bantignies, J. Martinez, M. Amblard. Turning peptide sequences into ribbon foldamers via a straightforward multi-cyclization reaction. Angew. Chem. Int. Ed., 2015, 54, 13966 –13970
B. Legrand, C. André, L. Moula, C. Didierjean, P. Hermet, J.L. Bantignies, J. Martinez, M. Amblard, M. Calmès, 12/14/14-Helix Formation in 2:1 α/β-Hybrid Peptides Containing Bicyclo[2.2.2]octane Ring Constraints, Chemistry: a European Journal, 2016, 22, 11986 –11990

Covalent‑reversible peptide‑based protease inhibitors. Design, synthesis, and clinical success stories.
Amino Acids 2023, https://link.springer.com/article/10.1007/s00726-023-03286-1
Feral, A. R. Martin, A. Desfoux, M. Amblard et L. Vezenkov.
Abstract
Dysregulated human peptidases are implicated in a large variety of diseases such as cancer, hypertension, and neurodegeneration. Viral proteases for their part are crucial for the pathogens’ maturation and assembly. Several decades of research were devoted to exploring these precious therapeutic targets, often addressing them with synthetic substrate-based inhibitors to elucidate their biological roles and develop medications. The rational design of peptide-based inhibitors offered a rapid pathway to obtain a variety of research tools and drug candidates. Non-covalent modifiers were historically the first choice for protease inhibition due to their reversible enzyme binding mode and thus presumably safer profile. However, in recent years, covalent-irreversible inhibitors are having a resurgence with dramatic increase of their related publications, preclinical and clinical trials, and FDA-approved drugs. Depending on the context, covalent modifiers could provide more effective and selective drug candidates, hence requiring lower doses, thereby limiting off-target effects. Additionally, such molecules seem more suitable to tackle the crucial issue of cancer and viral drug resistances. At the frontier of reversible and irreversible based inhibitors, a new drug class, the covalent-reversible peptide-based inhibitors, has emerged with the FDA approval of Bortezomib in 2003, shortly followed by 4 other listings to date. The highlight in the field is the breathtakingly fast development of the first oral COVID-19 medication, Nirmatrelvir. Covalent-reversible inhibitors can hipothetically provide the safety of the reversible modifiers combined with the high potency and specificity of their irreversible counterparts. Herein, we will present the main groups of covalent-reversible peptide-based inhibitors, focusing on their design, synthesis, and successful drug development programs.
The Unexpected Helical Supramolecular Assembly of a Simple Achiral Acetamide Tecton Generates Selective Water Channels
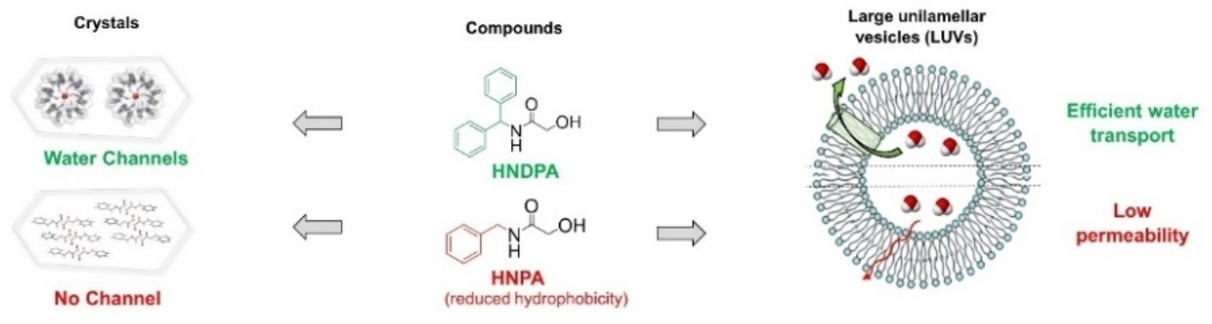
Abstract
Achiral 2-hydroxy-N-(diphenylmethyl)acetamide (HNDPA) crystallizes in the P61 chiral space group as a hydrate, building up permeable chiral crystalline helical water channels. The crystallization-driven chiral self-resolution process is highly robust, with the same air-stable crystalline form readily obtained under a variety of conditions. Interestingly, the HNDPA supramolecular helix inner pore is filled by a helical water wire. The whole edifice is mainly stabilized by robust hydrogen bonds involving the HNDPA amide bonds and CH… π interactions between the HNDPA phenyl groups. The crystalline structure shows breathing behavior, with completely reversible release and re-uptake of water inside the chiral channel under ambient conditions. Importantly, the HNDPA channel is able to transport water very efficiently and selectively under biomimetic conditions. With a permeability per channel of 3.3 million water molecules per second in large unilamellar vesicles (LUV) and total selectivity against NaCl, the HNDPA channel is a very promising functional nanomaterial for future applications.

1-Aminobicyclo[2.2.2]octane-2-carboxylic Acid and Derivatives As Chiral Constrained Bridged Scaffolds for Foldamers and Chiral Catalysts
Acc Chem Res. 2021 Feb 2;54(3):685-696. doi: 10.1021/acs.accounts.0c00680. Epub 2021 Jan 19.
Milbeo P, Martinez J, Amblard M, Calmès M, Legrand B
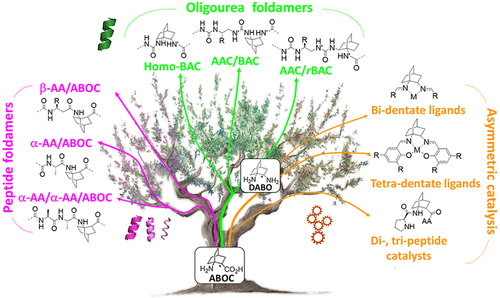
Abstract
The improvement of molecular diversity is one of the major concerns of chemists since the continuous development of original synthetic molecules provides unique scaffolds usable in organic and bioorganic chemistry. The challenge is to develop versatile platforms with highly controlled chemical three-dimensional space thanks to controlled chirality and conformational restraints. In this respect, cyclic β-amino acids are of great interest with applications in various fields of chemistry. In addition to their intrinsic biological properties, they are important precursors for the synthesis of new generations of bioactive compounds such as antibiotics, enzyme inhibitors, and antitumor agents. They have also been involved in asymmetric synthesis as efficient organo-catalysts in their free form and as derivatives. Finally, constrained cyclic β-amino acids have been incorporated into oligomers to successfully stabilize original structures in foldamer science with recent successes in health, material science, and catalysis. Over the last ∼10 years, we focused on bicyclic β-amino acids possessing a bicyclo[2.2.2]octane structure. This latter is a structural key element in numerous families of biologically active natural and synthetic products and is an interesting template for asymmetric synthesis. Nonetheless, reported studies on bicyclic carbo-bridged compounds are rather limited compared to those on bicyclic-fused and heterobridged derivatives. In this Account, we particularly focused on the synthesis and applications of the 1-aminobicyclo[2.2.2]octane-2-carboxylic acid, named, ABOC, and its derivatives. This highly constrained bicyclic β-amino acid, with a sterically hindered bridgehead primary amine and an endocyclic chiral center, displays drastically reduced conformational freedom. In addition, its high bulkiness strongly impacts the spatial orientation of the appended functionalities and the conformation of adjacent building blocks. Thus, we have first expanded a fundamental synthetic work by a wide ranging study in the field of foldamers, in the design of various stable peptide/peptidomimetic helical structures incorporating the ABOC residue (11/9-, 18/16-, 12/14/14-, and 12/10-helices). In addition, such bicyclic residue was fully compatible with and stabilized the canonical oligourea helix, whereas very few cyclic β-amino acids have been incorporated into oligoureas. In addition, we have pursued with the synthesis of some ABOC derivatives, in particular the 1,2-diaminobicyclo[2.2.2]octane chiral diamine, named DABO, and its investigation in chiral catalytic systems. Covalent organo-catalysis of the aldol reaction using ABOC-containing tripeptide catalysts provided a range of aldol products with high enantioselectivity. Moreover, the double reductive condensation of DABO with various aldehydes allowed the building of new chiral ligands that proved their efficiency in the copper-catalyzed asymmetric Henry reaction.
Potent Lys Patch-Containing Stapled Peptides Targeting PCSK9
J Med Chem. 2021 Aug 12;64(15):10834-10848. doi: 10.1021/acs.jmedchem.0c02051. Epub 2021 Jul 15.
Bourbiaux K, Legrand B, Verdié P, Mallart S, Manette G, Minoletti C, Stepp JD, Prigent P, Le Bail JC
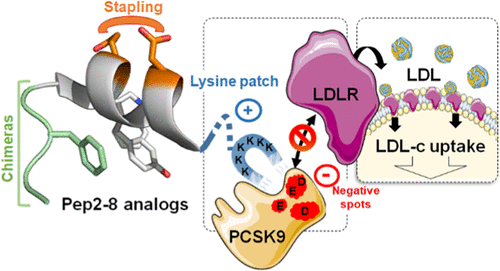
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9), identified as a regulator of low-density lipoprotein receptor (LDLR), plays a major role in cardiovascular diseases (CVD). Recently, Pep2-8, a small peptide with discrete three-dimensional structure, was found to inhibit the PCSK9/LDLR interaction. In this paper, we describe the modification of this peptide using stapled peptide and SIP technologies. Their combination yielded potent compounds such as 18 that potently inhibited the binding of PCSK9 to LDLR (KD = 6 ± 1 nM) and restored in vitro LDL uptake by HepG2 cells in the presence of PCSK9 (EC50 = 175 ± 40 nM). The three-dimensional structures of key peptides were extensively studied by circular dichroism and nuclear magnetic resonance, and molecular dynamics simulations allowed us to compare their binding mode to tentatively rationalize structure-activity relationships (SAR).
Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery.
Nanomaterials (Basel). 2020 Nov 25;10(12):2334. doi: 10.3390/nano10122334
Simon M, Laroui N, Heyraud M, Laconde G, Ali LMA, Bourbiaux K, Subra G, Vezenkov LL, Legrand B, Amblard M, Bettache N.
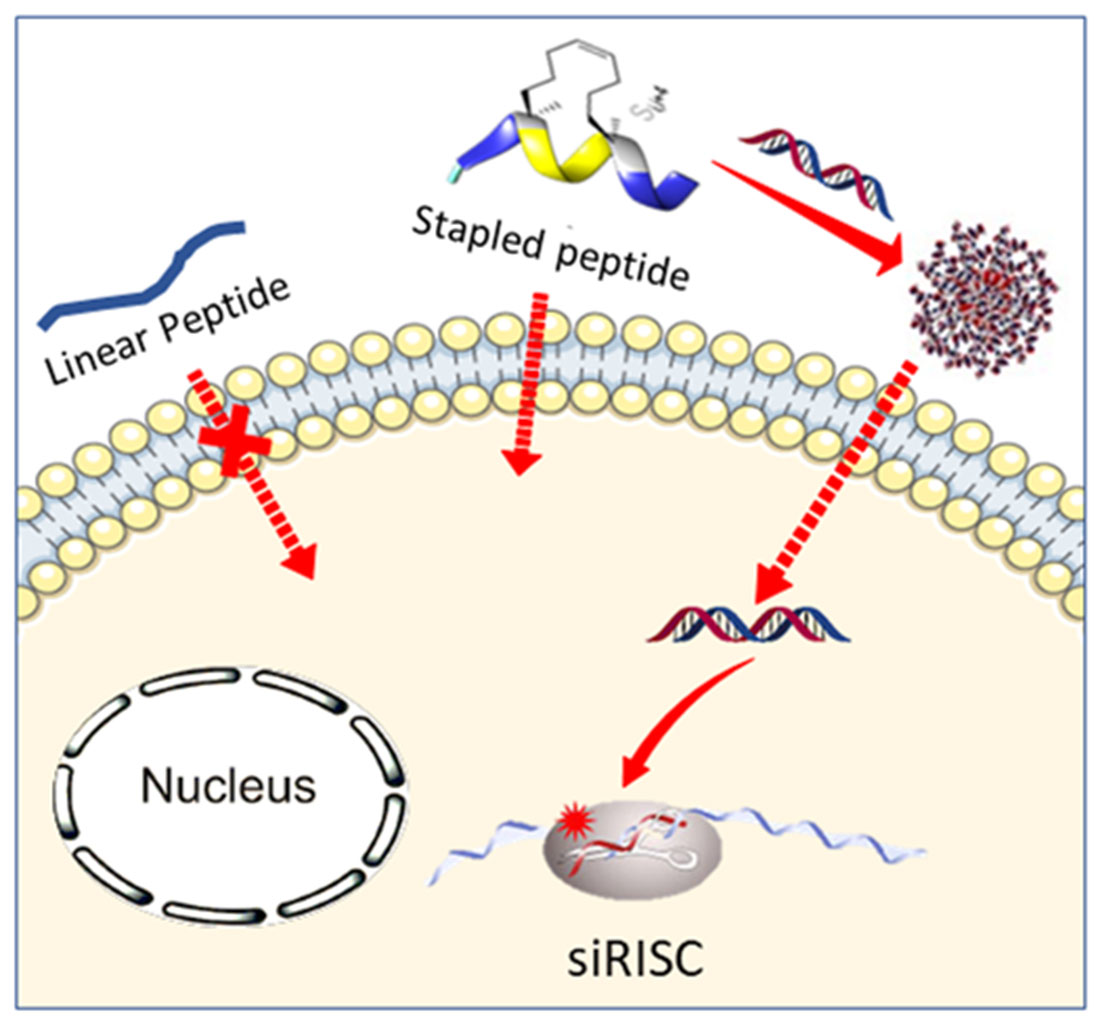
Abstract
Small interfering RNAs (siRNAs) are promising molecules for developing new therapies based on gene silencing; however, their delivery into cells remains an issue. In this study, we took advantage of stapled peptide technology that has emerged as a valuable strategy to render natural peptides more structured, resistant to protease degradation and more bioavailable, to develop short carriers for siRNA delivery. From the pool of stapled peptides that we have designed and synthesized, we identified non-toxic vectors that were able to efficiently encapsulate siRNA, transport them into the cell and induce gene silencing. Remarkably, the most efficient stapled peptide (JMV6582), is composed of only eight amino-acids and contains only two cationic charges.
Bottom-up strategies for the synthesis of peptide-based polymers
Progress in Polymer Science 115 (2021) 101377 https://doi.org/10.1016/j.progpolymsci.2021.101377
Martin J, Desfoux A, Martinez J, Amblard M, Mehdi A, Vezenkov L, Subra G
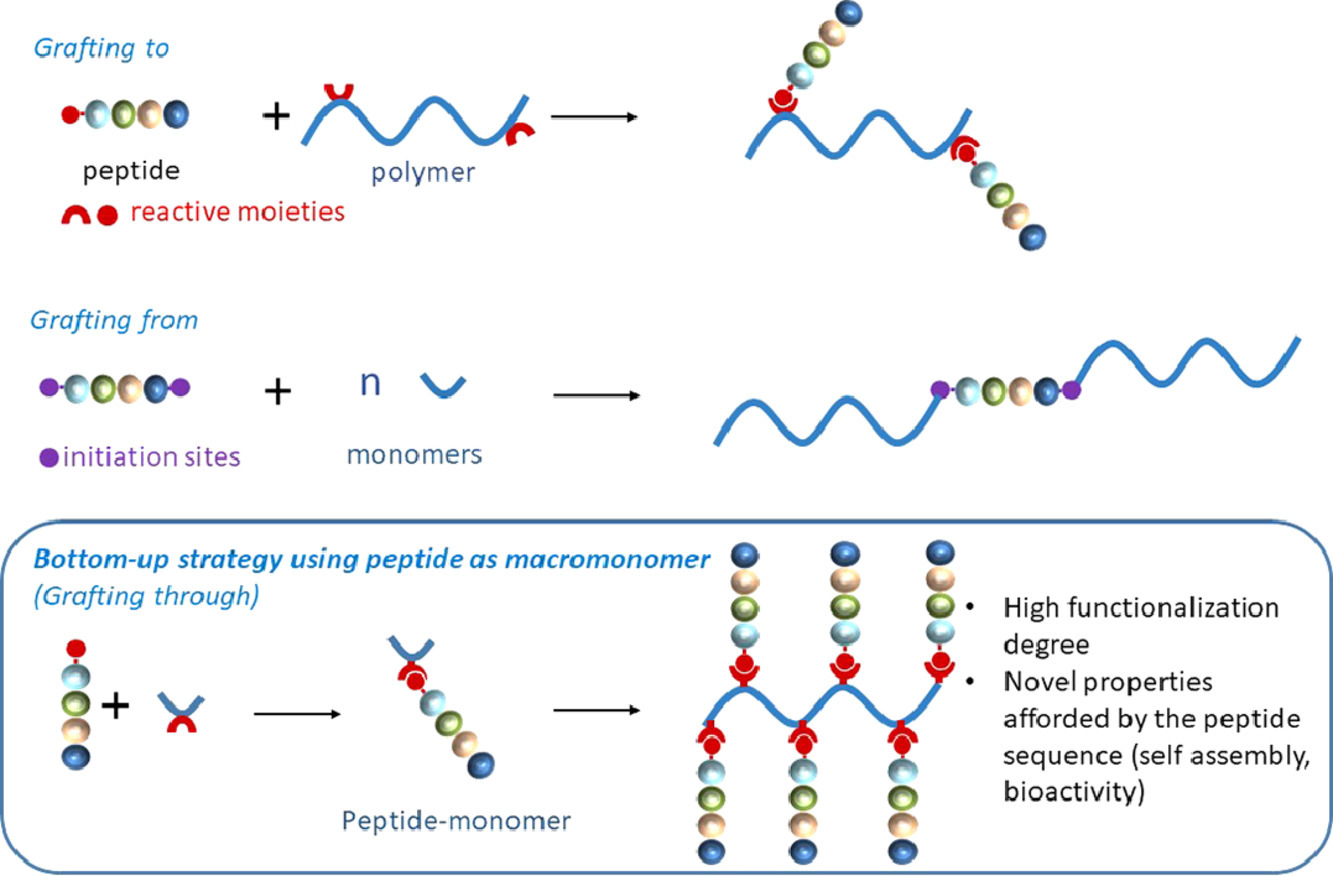
Abstract
Thanks to their wide range of biological activities, peptides have been extensively used to afford designed materials with tailored properties. Peptides can be associated to polymers combining the properties of various polymer backbones with those of bioactive peptide sequences. Such conjugates find promising applications in medical devices, tissue engineering, drug targeting and delivery. Improvement of existing polymers by post-modification peptide grafting is achieved through an extensive range of organic reactions, involving the prior preparation of functional polymers displaying suitable anchoring functions. Alternatively, peptides can be used as initiators of polymerization yielding a chimeric molecule bearing a single peptide at the end of macromolecular chains. Finally, novel polymer materials can be designed when the peptide itself is used as a macromonomer. In that case, the unmatched level of repetition of the peptide sequence or/and its self-assembly properties allow to access very high functionalization degree, original structures and bioactivities.
PROteolysis TArgetting Chimeras (PROTACs) Strategy Applied to Kinases: Recent Advances
Adv. Therap.2020, 2000148. https://doi.org/10.1002/adtp.202000148

Abstract
Since the development of the first protein kinase inhibitor in the early 1980s, followed by the FDA approval of imatinib in 2001, kinases are one of the most intensively pursued targets in current medicinal chemistry research. These proteins are overrepresented in various diseases such as cancer, inflammation or autoimmune pathologies and play important roles in their physiopathogenic processes. Despite the development and approval of numerous potent kinase inhibitors, drug resistance and off‐target side effects are commonly encountered with kinase inhibitors. Thus, development of novel strategies to overcome these problems is necessary. Since 2013, many research groups have proposed the conversion of potent kinase inhibitors into PROteolysis TArgeting Chimera (PROTAC) compounds and shared relevant and encouraging results using this new technology, which degrades proteins by employing the cellular machinery. Generally, this strategy brings enhancements in biological effects compared to the use of only the parent inhibitor. In this review article, recent findings related to the PROTAC technology applied to kinases are discussed, with a special focus on publications since 2018.
Self-Assembling Peptide—Polymer Nano-Objects via Polymerization-Induced Self-Assembly
Macromolecules 2020, 53, 16, 7034–7043 https://doi.org/10.1021/acs.macromol.0c01260
Dao T, Vezenkov L, Subra G, Amblard M, In M, Le Meins J-F, Aubrit F, Moradi M-A, Ladmiral A, Semsarilar M*
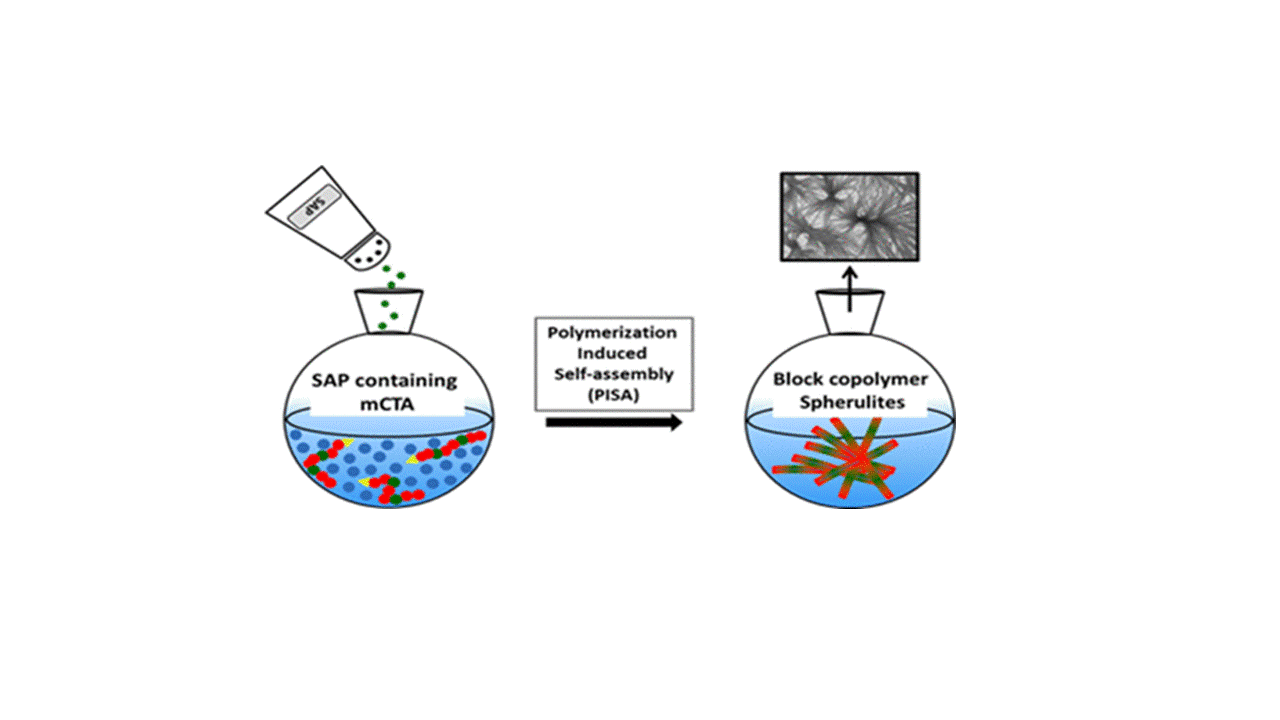
Abstract
Self-assembling peptides (SAPs) have been extensively studied for their ability to form nanoscale ordered structures driven by noncovalent molecular interactions. Meanwhile, polymerization-induced self-assembly (PISA) has been exploited as a facile and efficient way to produce various amphiphilic block copolymer nano-objects, whose self-assembly was governed predominantly by the interactions of the different blocks with the polymerization medium. In this work, we combined PISA with SAPs to prepare novel peptide–polymer hybrid nano-objects, thus harnessing the advantages of PISA and the self-assembling driving force of SAPs. A tripeptide methacrylamide derivative (MAm-Gly-Phe-Phe-NH2, denoted as MAm-GFF, where MAm means methacrylamide) was copolymerized with glycerol monomethacrylate (GMA) to produce a P(GMA65–stat-(MAm-GFF)7) macro-chain transfer agent (macro-CTA) by reversible addition–fragmentation chain transfer polymerization in dimethylformamide. This peptide-based macro-CTA was then successfully chain-extended with poly(2-hydroxypropyl methacrylate) (PHPMA) by aqueous dispersion PISA, forming P(GMA65–stat-(MAm-GFF)7)-b-PHPMA28 self-assembled objects. Fibrous structures were observed by transmission electron microscopy (TEM) and Cryo-TEM, in agreement with depolarized dynamic light scattering, static light scattering, and small-angle X-ray scattering experiments that also revealed long anisotropic morphologies. Such structures have not been reported previously for PISA-prepared nano-objects. This confirms the decisive influence of the GFF SAP on the self-assembly. In addition, annealing the PISA suspension at different temperatures led to a significant size decrease in the self-assembled objects and to a morphological transition caused by the thermosensitivity of both the core-forming PHPMA block and the stabilizing P(GMA-stat-(MAm-GFF)) block.
Inorganic Sol–Gel Polymerization for Hydrogel Bioprinting.
ACS Omega 2020, 5, 6, 2640-2647. doi: 10.1021/acsomega.9b03100.
Montheil T, Maumus M, Valot L, Lebrun A, Martinez J, Amblard M, Noël D, Mehdi A, Subra G.
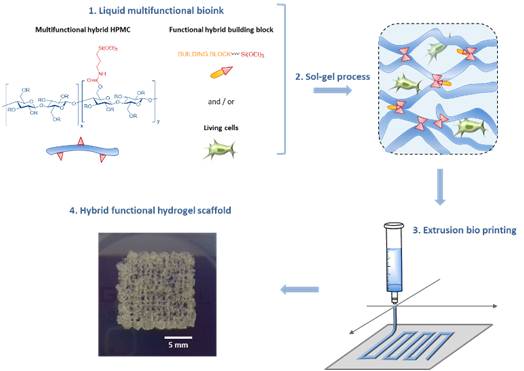
Abstract
An inorganic sol–gel polymerization process was used as a cross-linking reaction during three-dimensional (3D) bioprinting of cell-containing hydrogel scaffolds. Hybrid hydroxypropyl methyl cellulose (HPMC), with a controlled ratio of silylation, was prepared and isolated as a 3D-network precursor. When dissolved in a biological buffer containing human mesenchymal stem cells, it yields a bioink that can be printed during polymerization by extrusion. It is worth noting that the sol–gel process proceeded at pH 7.4 using biocompatible mode of catalysis (NaF and glycine). The printing window was determined by rheology and viscosity measurements. The physicochemical properties of hydrogels were studied. Covalent functionalization of the network can be easily performed by adding a triethoxysilyl-containing molecule; a fluorescent hybrid molecule was used as a proof of concept.
Self-mineralization and assembly of a bis-silylated Phe–Phe pseudodipeptide to a structured bioorganic–inorganic material
Mater. Horiz., 2019, 6, 2040-2046 doi: 10.1039/C9MH00580C
Jebors S, Valot L, Echalier C, Legrand L, Mikhaleff R,Van Der Lee A, Arenal R, Dumy P, Amblard M, Martinez J, Mehdi A and Subra G.
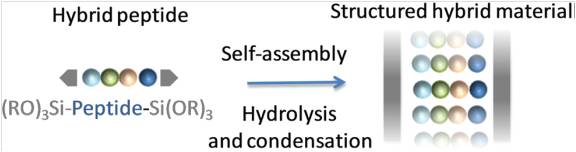
Abstract
Self-mineralization of a trialkoxysilyl hybrid peptide yields in a single step a nanostructured hybrid material. A bis-silylated pseudodipeptide inspired from the Phe–Phe dipeptide was used to program the assembly by sol–gel polymerization under heterogeneous conditions, in water at pH 1.5 without any structure-directing agent. A mechanism deciphering the hybrid material assembly was proposed thanks to 1H NMR spectroscopy. First, water-insoluble hybrid building blocks were hydrolysed into their soluble silanol counterparts. Then, these transitional species, thanks to hydrogen bonding and π–π stacking, self-assembled in solution. Last, the proximity of the silanol moieties favoured their polycondensation into growing siloxane oligomers, which spontaneously precipitated to produce an ordered hybrid material.
