
Jean-françois hernandez
Research Director (CNRS)
Jean-François Hernandez performed his PhD in the laboratory of Prof. Bernard P. Roques (Paris V University, 1987) under the supervision of Prof. Marie-Claude Fournié-Zaluski, where he developed pseudo-dipeptidic inhibitors of enkephalin-degrading enzymes as potential analgesics. Then, he went to the Salk Institute in San Diego as a post-doctoral fellow (1988-1990) under the supervision of Prof. Jean E. Rivier, where he learned solid phase peptide synthesis (Boc and Fmoc chemistries, GRF agonists, CRF antagonists, conotoxins, long peptides).
After moving back to France, he obtained a two-years temporary assistant professor position (ATER, 1990-1992) in the laboratory of Prof. Joëlle Paris at the faculty of Pharmacy of Lyon, where he developed SPPS methodology and worked on a project related to immunology. He obtained a permanent research position at the CNRS in 1992 and joined the team of Gérard Arlaud at Institut de Biologie Structurale in Grenoble, where he mainly performed the solid phase synthesis of peptides for fundamental and/or structural studies. He also contributed to the characterization of natural peptidase inhibitors (squash trypsin inhibitors like MCoTI) and of microbial peptidases.
In 2001, he moved to the laboratory of Prof. Jean Martinez (Laboratoire des Amino acides, Peptides et Protéines) in Montpellier, where he started to supervise a small group devoted to the development of enzyme inhibitors (pseudo-peptides, heterocycles) with therapeutical interest. Among targets that were or are currently explored: parasitic peptidases (Plasmodium, Leishmania), beta-lactamases, NO Synthases, Cathepsin D, secretases. A second important part of its activity is the development of solid phase synthetic strategies in the peptide/pseudopeptide field. In particular, he developed several strategies for the preparation of arginine- and arginine-like-containing compounds.
Currently, his activity mainly focuses on: (i) the development of heterocyclic molecules as inhibitors of metallo-β-lactamases, which are bacterial enzymes responsible of their resistance to carbapenem antibiotics, a major threat to human health; (ii) the development of pseudo-peptides inhibiting a peptidase of the Malaria-causing parasites Plasmodium falciparum and vivax.
Contact:
jean-francois.hernandez@umontpellier.fr
+33 (0)4 48 79 21 84
5 major publications :
Verdirosa F., Gavara L., Sevaille L., Tassone G., Corsica G., Legru A., Feller G., Chelini G., Mercuri P. S., Tanfoni S., Sannio F., Benvenuti M., Cerboni G., De Luca F., Bouajila E., Vo Hoang Y., Licznar-Fajardo P., Galleni M., Pozzi C., Mangani S., Docquier J.-D. & Hernandez J.-F. (2022) 1,2,4-Triazole-3-thione analogues with a 2-ethylbenzoic acid at position 4 as VIM-type metallo-β-lactamase inhibitors. ChemMedChem 17, e202100699.
Singh P., Samanta K., Kebe N. M., Michel G., Legrand B., Sitnikova V. E., Kajava A., Pagès M., Bastien P., Pomares C., Coux O. & Hernandez J.-F. (2022) The C-terminal segment of Leishmania major HslU: toward potential inhibitors of HslVU activity. Bioorg. Chem. 119, 105539.
Touati-Jallabe Y., Tintillier T., Mauchauffée E., Boucher J.-L., Leroy J., Ramassamy B., Hamzé A., Mezghenna K., Bouzekrini A., Verna C., Martinez J., Lajoix A.-D. & Hernandez J.-F. (2020) Solid phase synthesis of substrate-based dipeptides and heterocyclic pseudo-dipeptides as potential NO Synthase inhibitors. ChemMedChem 15, 517-531.
Gavara L., Sevaille L., De Luca F., Mercuri P., Bebrone C., Feller G., Legru A., Cerboni G., Tanfoni S., Baud D., Cutolo G., Bestgen B., Chelini G., Verdirosa F., Sannio F., Pozzi C., Benvenuti M., Kwapien K., Fischer M., Becker K., Frère J.-M., Mangani S., Gresh N., Berthomieu D., Galleni M., Docquier J.-D. & Hernandez J.-F. (2020) 4-Amino-1,2,4-triazole-3-thione-derived Schiff bases as metallo-β-lactamase inhibitors. Eur. J. Med. Chem. 208:112720.
Sevaille L., Gavara L., Bebrone C., De Luca F., Nauton L., Achard M., Mercuri P., Tanfoni S., Borgianni L., Guyon C., Lonjon P., Turan-Zitouni G., Dzieciolowski J., Becker K., Bénard L., Condon C., Maillard L., Martinez J., Frère J.-M., Dideberg O., Galleni M., Docquier J.-D. & Hernandez J.-F. (2017) 1,2,4-Triazole-3-thione compounds as inhibitors of di-zinc metallo-β-lactamases. ChemMedChem 12, 972-985.

The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides
Int J Mol Sci. 2019 Feb 26;20(5). pii: E1021. doi: 10.3390/ijms20051021
Kebe NM, Samanta K, Singh P, Lai-Kee-Him J, Apicella V, Payrot N, Lauraire N, Legrand B, Lisowski V, Mbang-Benet DE, Pages M, Bastien P, Kajava AV, Bron P, Hernandez JF, Coux O
Abstract
HslVU is an ATP-dependent proteolytic complex present in certain bacteria and in the mitochondrion of some primordial eukaryotes, including deadly parasites such as Leishmania. It is formed by the dodecameric protease HslV and the hexameric ATPase HslU, which binds via the C-terminal end of its subunits to HslV and activates it by a yet unclear allosteric mechanism. We undertook the characterization of HslV from Leishmania major (LmHslV), a trypanosomatid that expresses two isoforms for HslU, LmHslU1 and LmHslU2. Using a novel and sensitive peptide substrate, we found that LmHslV can be activated by peptides derived from the C-termini of both LmHslU1 and LmHslU2. Truncations, Ala- and D-scans of the C-terminal dodecapeptide of LmHslU2 (LmC12-U2) showed that five out of the six C-terminal residues of LmHslU2 are essential for binding to and activating HslV. Peptide cyclisation with a lactam bridge allowed shortening of the peptide without loss of potency. Finally, we found that dodecapeptides derived from HslU of other parasites and bacteria are able to activate LmHslV with similar or even higher efficiency. Importantly, using electron microscopy approaches, we observed that the activation of LmHslV was accompanied by a large conformational remodeling, which represents a yet unidentified layer of control of HslV activation.
1,2,4-Triazole-3-thione Compounds as Inhibitors of Dizinc Metallo-β-lactamases
ChemMedChem. 2017 Jun 21;12(12):972-985. doi: 10.1002/cmdc.201700186. Epub 2017 Jun 12.
Sevaille L, Gavara L, Bebrone C, De Luca F, Nauton L, Achard M, Mercuri P, Tanfoni S, Borgianni L, Guyon C, Lonjon P, Turan-Zitouni G, Dzieciolowski J, Becker K, Bénard L, Condon C, Maillard L, Martinez J, Frère JM, Dideberg O, Galleni M, Docquier JD, Hernandez JF.
Abstract
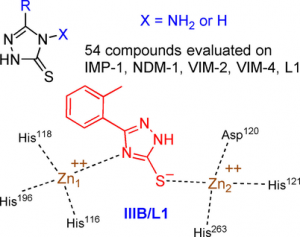
Metallo-β-lactamases (MBLs) cause resistance of Gram-negative bacteria to β-lactam antibiotics and are of serious concern, because they can inactivate the last-resort carbapenems and because MBL inhibitors of clinical value are still lacking. We previously identified the original binding mode of 4-amino-2,4-dihydro-5-(2-methylphenyl)-3H-1,2,4-triazole-3-thione (compound IIIA) within the dizinc active site of the L1 MBL. Herein we present the crystallographic structure of a complex of L1 with the corresponding non-amino compound IIIB (1,2-dihydro-5-(2-methylphenyl)-3H-1,2,4-triazole-3-thione). Unexpectedly, the binding mode of IIIB was similar but reverse to that of IIIA. The 3 D structures suggested that the triazole-thione scaffold was suitable to bind to the catalytic site of dizinc metalloenzymes. On the basis of these results, we synthesized 54 analogues of IIIA or IIIB. Nineteen showed IC50 values in the micromolar range toward at least one of five representative MBLs (i.e., L1, VIM-4, VIM-2, NDM-1, and IMP-1). Five of these exhibited a significant inhibition of at least four enzymes, including NDM-1, VIM-2, and IMP-1. Active compounds mainly featured either halogen or bulky bicyclic aryl substituents. Finally, some compounds were also tested on several microbial dinuclear zinc-dependent hydrolases belonging to the MBL-fold superfamily (i.e., endonucleases and glyoxalase II) to explore their activity toward structurally similar but functionally distinct enzymes. Whereas the bacterial tRNases were not inhibited, the best IC50 values toward plasmodial glyoxalase II were in the 10 μm range.
N-acyl benzotriazole derivatives for the synthesis of dipeptides and tripeptides and peptide biotinylation by mechanochemistry
ACS Sustainable Chemistry & Engineering, Pages: Ahead of Print, 2017, DOI: 10.1021/acssuschemeng.6b02439
L. Gonnet, T. Tintillier, N. Venturini, L. Konnert, J. F. Hernandez, F. Lamaty, G. Laconde, J. Martinez, E. Colacino
Abstract
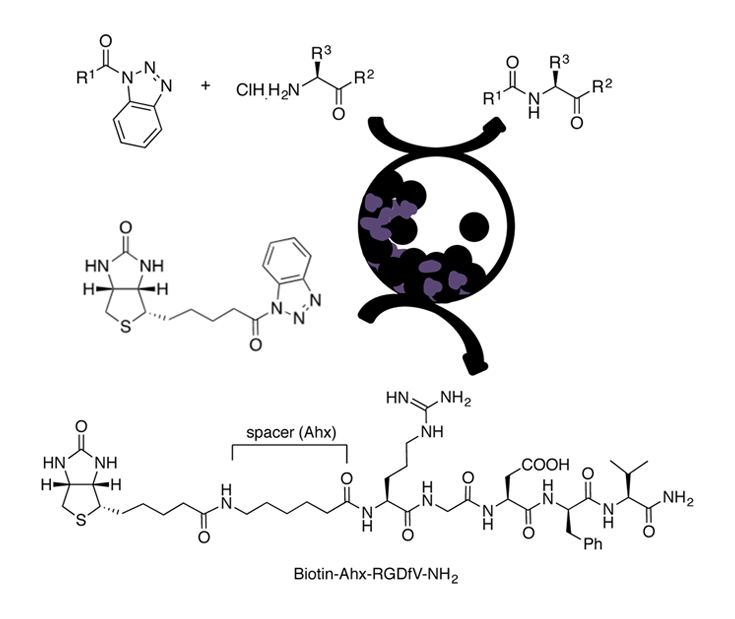
An eco-friendly methodol. for prepg. Fmoc-, Z-, and Boc-N-protected dipeptides and tripeptides is described, from the corresponding N-protected-α-aminoacyl benzotriazoles and α-amino acid derivs., with different C-terminal functionalities such as esters or amides, using vibrational ball-mill (VBM). The reactivity of a β-amino ester was also investigated. In some cases, the coupling was achieved by liq.-assisted grinding (LAG). α,α- and one α,β-dipeptide were obtained in good to excellent yields mainly by pptn. in water, resulting in an improved environmental impact compared to classical peptide synthesis in soln., as shown by green metric calcns. The method was extended to the biotinylation, via an aminohexanoyl spacer, of the pentapeptide RGDfV, which contains the well-known integrins recognition site arginine-glycine-aspartic acid (RGD) motif.
Structure-Activity Relationships of JMV4463, a Vectorized Cathepsin D Inhibitor with Antiproliferative Properties: The Unique Role of the AMPA-Based Vector
ChemMedChem, 2016, Volume: 11, Issue: 3, Pages: 302-308, DOI: 10.1002/cmdc.201500457
L. Vezenkov, C. A. Sanchez, V. Bellet, V. Martin, M. Maynadier, N. Bettache, V. Lisowski, J. Martinez, M. Garcia, M. Amblard, J. F. Hernandez
Abstract
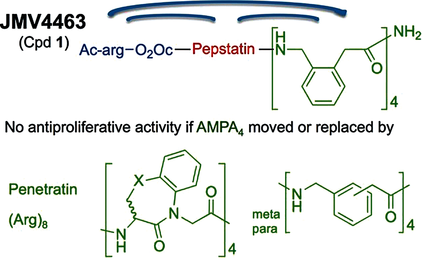
Cathepsin D (CathD) is overexpressed and secreted by several solid tumors and stimulates their growth, the mechanism of which is still not understood. In this context, the pepstatin bioconjugate JMV4463 [Ac-arg-O2Oc-(Val)3-Sta-Ala-Sta-(AMPA)4-NH2; O2Oc=8-amino-3,6-dioxaoctanoyl, Sta=statine, AMPA=ortho-aminomethylphenylacetyl], contg. a new kind of cell-penetrating vector, was previously shown to exhibit potent antiproliferative effects in vitro and to delay the onset of tumors in vivo. In this study, the authors performed a structure-activity relationship anal. to evaluate the significance of the inhibitor and vector moieties of JMV4463. By modifying both statine residues of pepstatin the authors found that the antiproliferative activity is correlated with CathD inhibition, supporting a major role of the catalytic activity of intracellular CathD in cancer cell proliferation. Replacing the vector composed of four AMPA units with other vectors was found to abolish cytotoxicity, although all of the conjugates enabled pepstatin transport into cells. In addn., the AMPA4 vector must be localized at the C terminus of the bioconjugate. The unexpected importance of the vector structure and position for cytotoxic action suggests that AMPA4 enables pepstatin to inhibit the proteolysis of crit. CathD substrates involved in cell proliferation via a unique mechanism of action.
Dipeptide mimic oligomer transporter mediates intracellular delivery of Cathepsin D inhibitors: a potential target for cancer therapy
J Control Release. 2013 Oct 28;171(2):251-7. doi: 10.1016/j.jconrel.2013.07.017. Epub 2013 Jul 27.
Maynadier M, Vezenkov LL, Amblard M, Martin V, Gandreuil C, Vaillant O, Gary-Bobo M, Basile I, Hernandez JF, Garcia M, Martinez J.
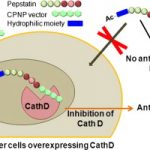
Abstract
Implication of the intracellular proteolytic activity of Cathepsin D (CathD), a lysosomal aspartyl-protease overexpressed in numerous solid tumors, has been evidenced on tumor growth. Its intracellular inhibition by potent inhibitors such as pepstatin constitutes a relevant but challenging molecular target. Indeed the potential of pepstatin as a therapeutic molecule is hampered by its too low intracellular penetration. We addressed this limitation by designing and developing a bioconjugate combining a pepstatin derivative with a new vector of cell penetration (CPNP) specifically targeting the endolysosomal compartment. We showed that this pepstatin conjugate (JMV4463) exhibited high anti-proliferative effect on tumor cell cultures via intracellular CathD inhibition and altered cell cycle associated with apoptotic events in vitro. When tested in mice xenografted with breast cancer cells, JMV4463 delayed tumor emergence and growth.
