Bioorg. Chem., 2022, 129, 106172, https://doi.org/10.1016/j.bioorg.2022.106172
R. El-Haggar, S. F. Hammad, R. I. Alsantali, M. M. Alrooqi, M. A. El Hassab, N. Masurier, M. F. Ahmed
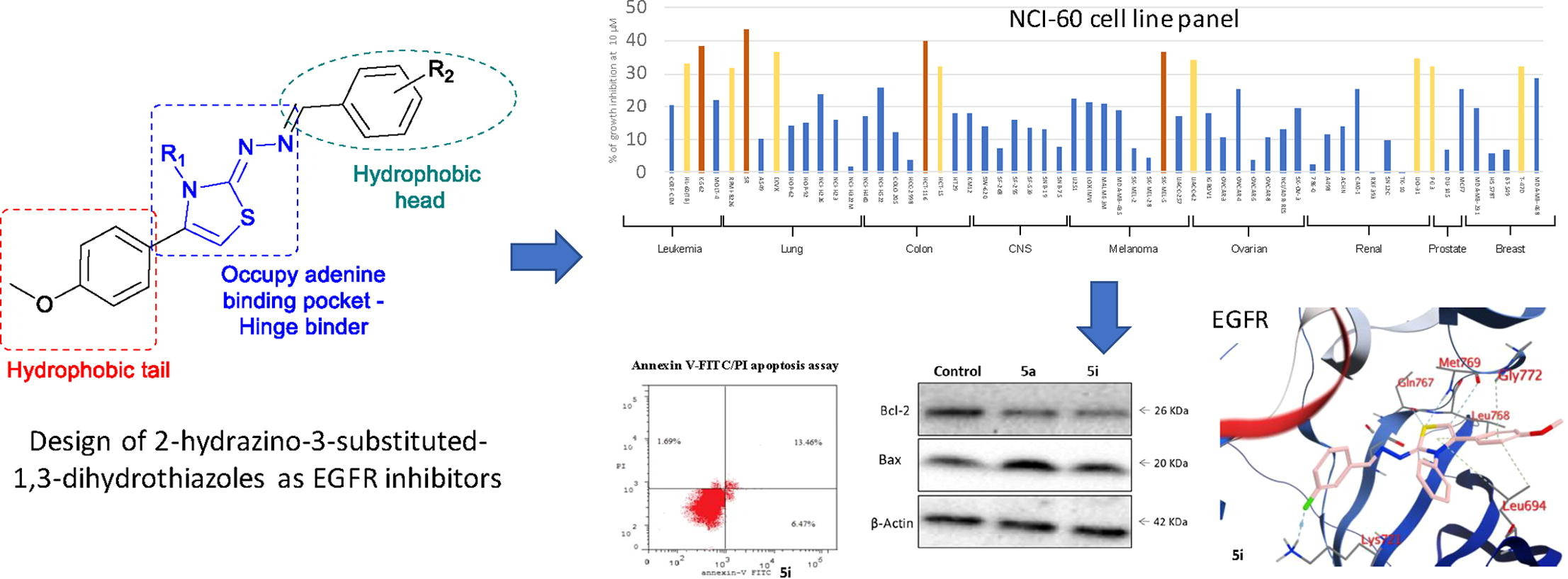
Abstract
The overexpression of EGFR has been recognized as the driver mechanism in the development of several human malignancies and the clinical use of EGFR inhibitors currently constitutes the standard of care for a wide range of malignancies, including colorectal cancer. However, the clinical efficacy of EGFR targeted inhibitors is limited by the development of intrinsic or acquired resistance, requiring the discovery of new compounds with different structural characteristics from those already developed. In this context, we explored the replacement of the aminoquinazoline pharmacophore of several FDA-approved EGFR inhibitors by its bioisosteric hydrazinothiazole moiety. A series of 14 new compounds were designed, synthesized, and evaluated as potential EGFR inhibitors. Compound 5i was active against 12 different cell lines in the NCI-60 cell line panel and showed an IC50 of 6.9 ± 0.013 µM against HCT-116 cells, with no significant toxicity against normal human fibroblasts (WI-38). Further studies showed that this compound showed submicromolar activity against EGFR and was able to induce tumor cell cycle arrest and cell apoptosis. Additionally, docking experiments, molecular dynamics and binding free energy calculations were performed and confirmed the potential of 2-hydrazino-2,3-dihydrothiazole derivatives as new EGFR inhibitors.
