
Jean-Alain Fehrentz
Research Director (CNRS)
Jean-Alain Fehrentz was born in Nancy, France, in 1955. He received his Ph.D. degree in Chemistry from the University of Nancy in 1983 and joined the Centre CNRS-INSERM de Pharmacologie Endocrinologie of Montpellier in the research group of Professor B. Castro. From 1989 to 1992 he was appointed as researcher at Sanofi Research in Montpellier. Then he moved to the Faculty of Pharmacy of Montpellier, working under the direction of Professor J. Martinez.
His research interests focus onpeptide aldehydes, enzyme inhibitors, peptidomimetics, and heterocycle based receptor ligands. He published more than 150 scientific papers.
Contact:
jean-alain.fehrentz(at)univ-montp1.fr
+33 411 75 96 06
5 major publications
L.S. Chatalic, M. Konijnenberg, J. Nonnekens, E. De Blois, S. Hoeben, C. De Ridder, L. Brunel, J. Martinez, J.-A. Fehrentz, D. C. Van Gent, B. A. Nock, T. Maina, W. M. Van Weerden, M. De Jong; In Vivo Stabilization of GRPR-peptide enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Theranostics 2016 6 (1), 104-117
G. P. Denis, A. Joly-Amado, E. Webber , F. Langlet,, M. Schaeffer, S. Padilla, C. Cansell, B. Dehouck, J. Castel, A.-S. Delbès, S.Martinez, A. Lacombe, C. Rouch, N. Kassis, J.-A. Fehrentz, J. Martinez, P. Verdié, T. S. Hnasko, R. D. Palmiter, M. Krashes, A. D. Güler, C. Magnan, S. Luquet; Palatability can drive feeding independent of AgRP neurons Cell Metab 2015 22, 4, 646-657
Damian, S. Mary, M. Maingot, C. M’Kadmi, D. Gagne, J-P. Leyris, S. Denoyelle, G. Gaibelet, L. Gavara, M. Garcia De Souza Costa, D. Perahia, E. Trinquet, B. Mouillac, S. Galandrin, C. Gales, J-A. Fehrentz, N. Floquet, J. Martinez, J. Marie, J-L. Baneres; Ghrelin receptor conformational dynamics regulate the transition from a preassembled to an active receptor:Gq complex. PNAS 2015 112 (5) 1601-1606 (2015)
Schaeffer, F. Langlet, C. Lafont, F. Molino, D. J. Hodson, T. Roux, L. Lamarque, P. Verdié, E. Bourrier, B. Dehouck, J.-L. Baneres, J. Martinez, P.-F. Méry, J. Marie, E. Trinquet, J.-A. Fehrentz, V. Prévot, P. Mollard; Rapid sensing of circulating ghrelin by hypothalamic appetite-modifying neurons. PNAS 2013 110(4) 1512-1517
J. -A. Fehrentz, B. Castro; An efficient synthesis of optically active -(t-butoxycarbonylamino)-aldehydes from -amino-acids. Synthesis 1983, vol 8, 676-679

Structure and dynamics of G protein-coupled receptor-bound ghrelin reveal the critical role of the octanoyl chain
Proc Natl Acad Sci U S A. 2019 Aug 27;116(35):17525-17530. doi: 10.1073/pnas.1905105116. Epub 2019 Aug 15
Ferré G, Louet M, Saurel O, Delort B, Czaplicki G, M’Kadmi C, Damian M, Renault P, Cantel S, Gavara L, Demange P, Marie J, Fehrentz JA, Floquet N, Milon A, Banères JL.
Abstract
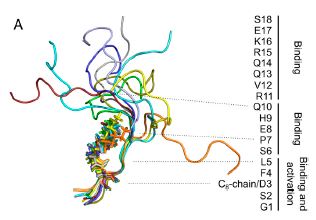
Ghrelin plays a central role in controlling major biological processes. As for other G protein-coupled receptor (GPCR) peptide agonists, the structure and dynamics of ghrelin bound to its receptor remain obscure. Using a combination of solution-state NMR and molecular modeling, we demonstrate that binding to the growth hormone secretagogue receptor is accompanied by a conformational change in ghrelin that structures its central region, involving the formation of a well-defined hydrophobic core. By comparing its acylated and nonacylated forms, we conclude that the ghrelin octanoyl chain is essential to form the hydrophobic core and promote access of ghrelin to the receptor ligand-binding pocket. The combination of coarse-grained molecular dynamics studies and NMR should prove useful in improving our mechanistic understanding of the complex conformational space explored by a natural peptide agonist when binding to its GPCR. Such information should also facilitate the design of new ghrelin receptor-selective drugs.
Synthesis of [1,2,4]Triazolo[4,3- a]piperazin-6-ones: An Approach to the Triazole-Fused Ketopiperazine Scaffold
Org Lett. 2018 Jun 1;20(11):3250-3254. doi: 10.1021/acs.orglett.8b01112. Epub 2018 May 15.
Ben Haj Salah K, Legrand B, Bibian M, Wenger E, Fehrentz JA, Denoyelle S.
Abstract
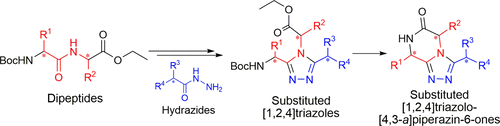
A stereoconservative synthesis to access the triazole-fused ketopiperazine (TKP) scaffold is presented. This underexplored platform offers a wide range of structural modulations with several points of diversity and chiral centers. A series of [1,2,4]triazolo[4,3- a]piperazin-6-ones was synthesized from optically pure dipeptides. The methodology was then successfully applied to access the pyrrolo[1,2- a]triazolo[3,4- c]piperazin-6-one tricycle. Importantly, the crystal structures of representative TKPs confirmed that the configuration of the chiral centers was controlled during the synthetic route and facilitated description of the orientation of the substituents depending on their nature and position on the TKP scaffold.
GHSR-D2R heteromerization modulates dopamine signaling through an effect on G protein conformation
Proc Natl Acad Sci U S A. 2018 Apr 24;115(17):4501-4506. doi: 10.1073/pnas.1712725115. Epub 2018 Apr 9
Damian M, Pons V, Renault P, M’Kadmi C, Delort B, Hartmann L, Kaya AI, Louet M, Gagne D, Ben Haj Salah K, Denoyelle S, Ferry G, Boutin JA, Wagner R, Fehrentz JA, Martinez J, Marie J, Floquet N, Galès C, Mary S, Hamm HE, Banères JL.
Abstract
IThe growth hormone secretagogue receptor (GHSR) and dopamine receptor (D2R) have been shown to oligomerize in hypothalamic neurons with a significant effect on dopamine signaling, but the molecular processes underlying this effect are still obscure. We used here the purified GHSR and D2R to establish that these two receptors assemble in a lipid environment as a tetrameric complex composed of two each of the receptors. This complex further recruits G proteins to give rise to an assembly with only two G protein trimers bound to a receptor tetramer. We further demonstrate that receptor heteromerization directly impacts on dopamine-mediated Gi protein activation by modulating the conformation of its α-subunit. Indeed, association to the purified GHSR:D2R heteromer triggers a different active conformation of Gαi that is linked to a higher rate of GTP binding and a faster dissociation from the heteromeric receptor. This is an additional mechanism to expand the repertoire of GPCR signaling modulation that could have implications for the control of dopamine signaling in normal and physiopathological conditions.
The GHR-R antagonist JMV 2959 neither induces malaise nor alters the malaise property of LiCl in the adult male rat
Physiol Behav. 2018 Jan 1;183:46-48. doi: 10.1016/j.physbeh.2017.10.017. Epub 2017 Oct 19.
Rodriguez JA, Fehrentz JA, Martinez J, Ben Haj Salah K, Wellman PJ.
Abstract
The orexigenic peptide ghrelin (GHR) interacts with ghrelin receptors (GHR-Rs) to modulate brain reinforcement and feeding circuits. Pharmacological inactivation of GHR-Rs via administration of the drug JMV 2959 attenuates the rewarding/reinforcing effects of several drugs of abuse including alcohol, morphine, amphetamine and nicotine. One view of these results is that inactivation of GHR-Rs taps into brain reinforcement/feeding circuits acted upon by drugs of abuse. An alternate explanation is that JMV 2959 may induce malaise, which in turn may limit reinforcement as well as food ingestion. This is a variable of interest given that nicotine alone can induce malaise which may be enhanced by JMV 2959. In the present study, we assessed the capacity of JMV 2959 to produce malaise using a conditioned taste aversion (CTA) task. Adult male rats were allowed to consume a 0.1% sodium saccharin solution and then injected IP with either vehicle, 0.4mg/kg nicotine, 3mg/kg JMV 2959, a combination of 0.4mg/kg nicotine and 3mg/kg JMV 2959, or 32mg/kg lithium chloride (a positive control known to support induction of CTA). Lithium chloride produced a robust avoidance of the saccharin solution in subsequent 2 bottle (water and saccharin) tests, whereas JMV 2959 alone did not induce CTA. The combination of JMV 2959 and nicotine induced a moderate degree of CTA that was similar to that produced by nicotine alone. These results suggest that JMV 2959 is unlikely to limit either reinforcement or food ingestion via induction of malaise.
Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia
Sci Rep. 2017 Oct 12;7(1):13017. doi: 10.1038/s41598-017-13504-y.
Sirago G, Conte E, Fracasso F, Cormio A, Fehrentz JA, Martinez J, Musicco C, Camerino GM, Fonzino A, Rizzi L, Torsello A, Lezza AMS, Liantonio A, Cantatore P, Pesce V.
Abstract
Chemotherapy can cause cachexia, which consists of weight loss associated with muscle atrophy. The exact mechanisms underlying this skeletal muscle toxicity are largely unknown and co-therapies to attenuate chemotherapy-induced side effects are lacking. By using a rat model of cisplatin-induced cachexia, we here characterized the mitochondrial homeostasis in tibialis anterior cachectic muscle and evaluated the potential beneficial effects of the growth hormone secretagogues (GHS) hexarelin and JMV2894 in this setting. We found that cisplatin treatment caused a decrease in mitochondrial biogenesis (PGC-1α, NRF-1, TFAM, mtDNA, ND1), mitochondrial mass (Porin and Citrate synthase activity) and fusion index (MFN2, Drp1), together with changes in the expression of autophagy-related genes (AKT/FoxO pathway, Atg1, Beclin1, LC3AII, p62) and enhanced ROS production (PRX III, MnSOD). Importantly, JMV2894 and hexarelin are capable to antagonize this chemotherapy-induced mitochondrial dysfunction. Thus, our findings reveal a key-role played by mitochondria in the mechanism responsible for GHS beneficial effects in skeletal muscle, strongly indicating that targeting mitochondrial dysfunction might be a promising area of research in developing therapeutic strategies to prevent or limit muscle wasting in cachexia.
Involvement of PPARγ in the Anticonvulsant Activity of EP-80317, a Ghrelin Receptor Antagonist
Front Pharmacol. 2017 Sep 22;8:676. doi: 10.3389/fphar.2017.00676. eCollection 2017.
Lucchi C, Costa AM, Giordano C, Curia G, Piat M, Leo G, Vinet J, Brunel L, Fehrentz JA, Martinez J, Torsello A, Biagini G.
Abstract
Ghrelin, des-acyl ghrelin and other related peptides possess anticonvulsant activities. Although ghrelin and cognate peptides were shown to physiologically regulate only the ghrelin receptor, some of them were pharmacologically proved to activate the peroxisome proliferator-activated receptor gamma (PPARγ) through stimulation of the scavenger receptor CD36 in macrophages. In our study, we challenged the hypothesis that PPARγ could be involved in the anticonvulsant effects of EP-80317, a ghrelin receptor antagonist. For this purpose, we used the PPARγ antagonist GW9662 to evaluate the modulation of EP-80317 anticonvulsant properties in two different models. Firstly, the anticonvulsant effects of EP-80317 were studied in rats treated with pilocarpine to induce status epilepticus (SE). Secondly, the anticonvulsant activity of EP-80317 was ascertained in the repeated 6-Hz corneal stimulation model in mice. Behavioral and video electrocorticographic (ECoG) analyses were performed in both models. We also characterized levels of immunoreactivity for PPARγ in the hippocampus of 6-Hz corneally stimulated mice. EP-80317 predictably antagonized seizures in both models. Pretreatment with GW9662 counteracted almost all EP-80317 effects both in mice and rats. Only the effects of EP-80317 on power spectra of ECoGs recorded during repeated 6-Hz corneal stimulation were practically unaffected by GW9662 administration. Moreover, GW9662 alone produced a decrease in the latency of tonic-clonic seizures and accelerated the onset of SE in rats. Finally, in the hippocampus of mice treated with EP-80317 we found increased levels of PPARγ immunoreactivity. Overall, these results support the hypothesis that PPARγ is able to modulate seizures and mediates the anticonvulsant effects of EP-80317.
Pharmacological and Biochemical Characterization of TLQP-21 Activation of a Binding Site on CHO Cells
Front Pharmacol. 2017, Mar 30;8:167. doi: 10.3389/fphar.2017.00167. eCollection 2017.
Molteni L, Rizzi L, Bresciani E, Possenti R, Petrocchi Passeri P, Ghè C, Muccioli G, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, Biagini G, Binda A, Rivolta I, Locatelli V, Torsello A.
Abstract
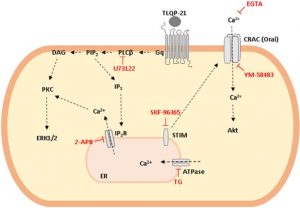
VGF is a propeptide of 617 amino acids expressed throughout the central and the peripheral nervous system. VGF and peptides derived from its processing have been found in dense core vesicles and are released from neuronal and neuroendocrine cells via the regulated secretory pathway. Among VGF-derived neuropeptides, TLQP-21 (VGF556-576) has raised a huge interest and is one of most studied. TLQP-21 is a multifunctional neuropeptide involved in the control of several physiological functions, potentially including energy homeostasis, pain modulation, stress responsiveness and reproduction. Although little information is available about its receptor and the intracellular mechanisms mediating its biological effects, recent reports suggest that TLQP-21 may bind to the complement receptors C3aR1 and/or gC1qR. The first aim of this study was to ascertain the existence and nature of TLQP-21 binding sites in CHO cells. Secondly, we endeavored to characterize the ligand binding to these sites by using a small panel of VGF-derived peptides. And finally, we investigated the influence of TLQP-21 on selected intracellular signaling pathways. We report that CHO cells express a single class of saturable and specific binding sites for TLQP-21 with an affinity and capacity of Kd = 0.55 ± 0.05 × 10-9 M and Bmax = 81.7 ± 3.9 fmol/mg protein, respectively. Among the many bioactive products derived from the C-terminal region of VGF that we tested, TLQP-21 was the most potent in stimulating intracellular calcium mobilization in CHO cells; this effect is primarily due to its C-terminal fragment (HFHH-10). TLQP-21 induced rapid and transient dephosphorylation of phospholipase Cγ1 and phospholipase A2. Generation of IP3 and diacylglycerol was crucial for TLQP-21 bioactivity. In conclusion, our results suggest that the receptor stimulated by TLQP-21 belongs to the family of the Gq-coupled receptors, and its activation first increases membrane-lipid derived second messengers which thereby induce the mobilization of Ca2+ from the endoplasmic reticulum followed by a slower store-operated Ca2+ entry from outside the cell.
Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia.
J Cachexia Sarcopenia Muscle. 2017, Jun;8(3):386-404. doi: 10.1002/jcsm.12185. Epub 2017 Mar 10.
Conte E, Camerino GM, Mele A, De Bellis M, Pierno S, Rana F, Fonzino A, Caloiero R, Rizzi L, Bresciani E, Ben Haj Salah K, Fehrentz JA, Martinez J, Giustino A, Mariggiò MA, Coluccia M, Tricarico D, Lograno MD, De Luca A, Torsello A, Conte D, Liantonio A.
Abstract
BACKGROUND:
Cachexia is a wasting condition associated with cancer types and, at the same time, is a serious and dose-limiting side effect of cancer chemotherapy. Skeletal muscle loss is one of the main characteristics of cachexia that significantly contributes to the functional muscle impairment. Calcium-dependent signaling pathways are believed to play an important role in skeletal muscle decline observed in cachexia, but whether intracellular calcium homeostasis is affected in this situation remains uncertain. Growth hormone secretagogues (GHS), a family of synthetic agonists of ghrelin receptor (GHS-R1a), are being developed as a therapeutic option for cancer cachexia syndrome; however, the exact mechanism by which GHS interfere with skeletal muscle is not fully understood.
METHODS:
By a multidisciplinary approach ranging from cytofluorometry and electrophysiology to gene expression and histology, we characterized the calcium homeostasis in fast-twitch extensor digitorum longus (EDL) muscle of adult rats with cisplatin-induced cachexia and established the potential beneficial effects of two GHS (hexarelin and JMV2894) at this level. Additionally, in vivo measures of grip strength and of ultrasonography recordings allowed us to evaluate the functional impact of GHS therapeutic intervention.
RESULTS:
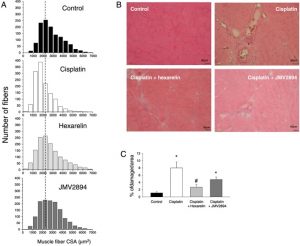
Cisplatin-treated EDL muscle fibres were characterized by a ~18% significant reduction of the muscle weight and fibre diameter together with an up-regulation of atrogin1/Murf-1 genes and a down-regulation of Pgc1-a gene, all indexes of muscle atrophy, and by a two-fold increase in resting intracellular calcium, [Ca2+ ]i , compared with control rats. Moreover, the amplitude of the calcium transient induced by caffeine or depolarizing high potassium solution as well as the store-operated calcium entry were ~50% significantly reduced in cisplatin-treated rats. Calcium homeostasis dysregulation parallels with changes of functional ex vivo (excitability and resting macroscopic conductance) and in vivo (forelimb force and muscle volume) outcomes in cachectic animals. Administration of hexarelin or JMV2894 markedly reduced the cisplatin-induced alteration of calcium homeostasis by both common as well as drug-specific mechanisms of action. This effect correlated with muscle function preservation as well as amelioration of various atrophic indexes, thus supporting the functional impact of GHS activity on calcium homeostasis.
CONCLUSIONS:
Our findings provide a direct evidence that a dysregulation of calcium homeostasis plays a key role in cisplatin-induced model of cachexia gaining insight into the etiopathogenesis of this form of muscle wasting. Furthermore, our demonstration that GHS administration efficaciously prevents cisplatin-induced calcium homeostasis alteration contributes to elucidate the mechanism of action through which GHS could potentially ameliorate chemotherapy-associated cachexia.
JMV5656, A Novel Derivative of TLQP-21, Triggers the Activation of a Calcium-Dependent Potassium Outward Current in Microglial Cells
Front Cell Neurosci. 2017 Feb 23;11:41. doi: 10.3389/fncel.2017.00041. eCollection 2017
Rivolta I, Binda A, Molteni L, Rizzi L, Bresciani E, Possenti R, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, Locatelli V, Torsello A.
Abstract
TLQP-21 (TLQPPASSRRRHFHHALPPAR) is a multifunctional peptide that is involved in the control of physiological functions, including feeding, reproduction, stress responsiveness, and general homeostasis. Despite the huge interest in TLQP-21 biological activity, very little is known about its intracellular mechanisms of action. In microglial cells, TLQP-21 stimulates increases of intracellular Ca2+ that may activate functions, including proliferation, migration, phagocytosis and production of inflammatory molecules. Our aim was to investigate whether JMV5656 (RRRHFHHALPPAR), a novel short analogue of TLQP-21, stimulates intracellular Ca2+ in the N9 microglia cells, and whether this Ca2+ elevation is coupled with the activation Ca2+-sensitive K+ channels. TLQP-21 and JMV5656 induced a sharp, dose-dependent increment in intracellular calcium. In 77% of cells, JMV5656 also caused an increase in the total outward currents, which was blunted by TEA (tetraethyl ammonium chloride), a non-selective blocker of voltage-dependent and Ca2+-activated potassium (K+) channels. Moreover, the effects of ion channel blockers charybdotoxin and iberiotoxin, suggested that multiple calcium-activated K+ channel types drove the outward current stimulated by JMV5656. Additionally, inhibition of JMV5656-stimulated outward currents by NS6180 (4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4 benzothiazin-3(4H)-one) and TRAM-34 (triarylmethane-34), indicated that KCa3.1 channels are involved in this JMV5656 mechanisms of action. In summary, we demonstrate that, in N9 microglia cells, the interaction of JMV5656 with the TLQP-21 receptors induced an increase in intracellular Ca2+, and, following extracellular Ca2+ entry, the opening of KCa3.1 channels.
JMV2894, a novel growth hormone secretagogue, accelerates body mass recovery in an experimental model of cachexia
Endocrine, 2016, Pages: Ahead of Print
E. Bresciani, L. Rizzi, L. Molteni, M. Ravelli, A. Liantonio, K. Ben Haj Salah, J. A. Fehrentz, J. Martinez, R. J. Omeljaniuk, G. Biagini, V. Locatelli, A. Torsello
Abstract
Oncol. patients subjected to chemotherapy frequently present aphagia, malnutrition, and cachexia. The purpose of this study was to investigate whether selected growth hormone secretagogues including hexarelin, JMV2894 and JMV2951 could antagonize body wt. loss and wasting induced by cisplatin administration in rats. The three growth hormone secretagogues behaved as full agonists of the growth hormone secretagogues receptor both in terms of ability to stimulate calcium mobilization in Chinese hamster ovary cells and stimulation of growth hormone release in neonatal rats. Adult rats were (i) treated with vehicle throughout (controls), or (ii) treated with cisplatin (days 1-3) and a growth hormone secretagogues or vehicle, (days 1-12). Body wt. and food consumption were measured daily. Although all growth hormone secretagogues caused initial transient acute increases in food intake, the total amt. of food eaten by controls and growth hormone secretagogues treated groups over the 12 exptl. days was not significantly different. All groups pre-treated with cisplatin lost up to 5-10 % body wt. in the first 4 days; they subsequently gained wt. at a rate comparable with controls. Interestingly, rats which received JMV2894 demonstrated a faster gain in body wt. than any other growth hormone secretagogues treated group and at the end of the protocol reached a wt. similar to that of controls. JMV2894 did not stimulate perirenal and epididymal fat accumulation but reduced MuRF mRNA levels in skeletal muscles. In conclusion, our findings demonstrate that JMV2894 antagonizes cisplatin induced wt. loss in rats and may prove useful in antagonizing cachexia assocd. with cancer and chemotherapy in humans.
