
Lubomir Vezenkov
ASSOCIATE Professor, National Graduate School of Chemistry of Montpellier
Lubomir Vezenkov was born in Bulgaria, where he performed his studies until he arrived in Montpellier in 2005 for his masters in the ENSCM engineering school.
Then, he performed a co-tutoring PhD between the universities of Montpellier and Naples, which he graduated from in 2011. During this period, he helped develop some potent cell penetrating foldamers that were used to vectorize pepststin and provide selective anti-cancer activity.
Afterwards, he did a two-years-long post-doctoral internship in the prof. Robert Young group in Simon Fraser University, Vancouver, where he took part in the development of one the first specific autophagy inhibitors.
Then, he moved back to Montpellier, and in 2016, he received an associate professor position at ENSCM in team 9 of the IBMM research institute.
Currently, he is working in the fields of peptide and medicinal chemistry as well as in the exciting domain of chemical biology. Recently he has been interested in the development of covalent-irreversible and covalent-reversible protease inhibitors. These covalent modifiers are used to block or activate certain microtubulin post-translational modifications. Such compounds can help better understand the polarization of neurons and can be used as therapeutics for neurodegenerative disease.
lubomir.vezenkov@enscm.fr
+33 448792185
5 major publications :
Dao T, Vezenkov L, Subra G, Amblard M, In M, Le Meins J-F, Aubrit F, Moradi M-A, Ladmiral V, Semsarilar M; Self-Assembling Peptide—Polymer Nano-Objects via Polymerization-Induced Self-Assembly, Macromolecules 2020, 53, 16, 7034–7043. https://doi.org/10.1021/acs.macromol.0c01260
Bosc D, Vezenkov L, Bortnik S, An J, Xu J, Choutka C, Hannigan AM, Kovacic S, Loo S, Clark PGK, Chen G, Guay-Ross RN, Yang K, Dragowska WH, Zhang F, Go NE, Leung A, Honson NS, Pfeifer TA, Gleave M, Bally M, Jones SJ, Gorski SM, Young RN; A new quinoline-based chemical probe inhibits the autophagy-related cysteine protease ATG4B, Sci Rep., 2018 Aug 3;8(1):11653. doi: 10.1038/s41598-018-29900-x.
Vezenkov L, Martin V, Bettache N, Simon M, Messerschmitt A, Legrand B, Bantignies JL, Subra G, Maynadier M, Bellet V, Garcia M, Martinez J, Amblard M; Ribbon-like Foldamers for Cellular Uptake and Drug Delivery, Chembiochem, 2017 Nov 2;18(21):2110-2114 https://doi.org/10.1002/cbic.201700455
Vezenkov L, Maynadier M, Amblard M, Martin V, Gandreuil C, Vaillant O, Gary-Bobo M, Basile I, Hernandez JF, Garcia M, Martinez J; Dipeptide mimic oligomer transporter mediates intracellular delivery of Cathepsin D inhibitors: a potential target for cancer therapy. Journal of Controlled Release, 2013 Oct 28;171(2):251-7. https://doi.org/10.1016/j.jconrel.2013.07.017Get rights and content
Vezenkov L, Maynadier M, Hernandez JF, Averlant-Petit M, Fabre O, Garcia M, Martinez J, Amblard M; Noncationic Dipeptide Mimic Oligomers As Cell Penetrating Nonpeptides(CPNP), Bioconjugate Chem. 2010, Sept 21(10), 1850–1854. https://doi.org/10.1021/bc1002086

Covalent‑reversible peptide‑based protease inhibitors. Design, synthesis, and clinical success stories.
Amino Acids 2023, https://link.springer.com/article/10.1007/s00726-023-03286-1
Feral, A. R. Martin, A. Desfoux, M. Amblard et L. Vezenkov.
Abstract
Dysregulated human peptidases are implicated in a large variety of diseases such as cancer, hypertension, and neurodegeneration. Viral proteases for their part are crucial for the pathogens’ maturation and assembly. Several decades of research were devoted to exploring these precious therapeutic targets, often addressing them with synthetic substrate-based inhibitors to elucidate their biological roles and develop medications. The rational design of peptide-based inhibitors offered a rapid pathway to obtain a variety of research tools and drug candidates. Non-covalent modifiers were historically the first choice for protease inhibition due to their reversible enzyme binding mode and thus presumably safer profile. However, in recent years, covalent-irreversible inhibitors are having a resurgence with dramatic increase of their related publications, preclinical and clinical trials, and FDA-approved drugs. Depending on the context, covalent modifiers could provide more effective and selective drug candidates, hence requiring lower doses, thereby limiting off-target effects. Additionally, such molecules seem more suitable to tackle the crucial issue of cancer and viral drug resistances. At the frontier of reversible and irreversible based inhibitors, a new drug class, the covalent-reversible peptide-based inhibitors, has emerged with the FDA approval of Bortezomib in 2003, shortly followed by 4 other listings to date. The highlight in the field is the breathtakingly fast development of the first oral COVID-19 medication, Nirmatrelvir. Covalent-reversible inhibitors can hipothetically provide the safety of the reversible modifiers combined with the high potency and specificity of their irreversible counterparts. Herein, we will present the main groups of covalent-reversible peptide-based inhibitors, focusing on their design, synthesis, and successful drug development programs.

Hydrocarbon-Stapled Peptide Based-Nanoparticles for siRNA Delivery.
Nanomaterials (Basel). 2020 Nov 25;10(12):2334. doi: 10.3390/nano10122334
Simon M, Laroui N, Heyraud M, Laconde G, Ali LMA, Bourbiaux K, Subra G, Vezenkov LL, Legrand B, Amblard M, Bettache N.
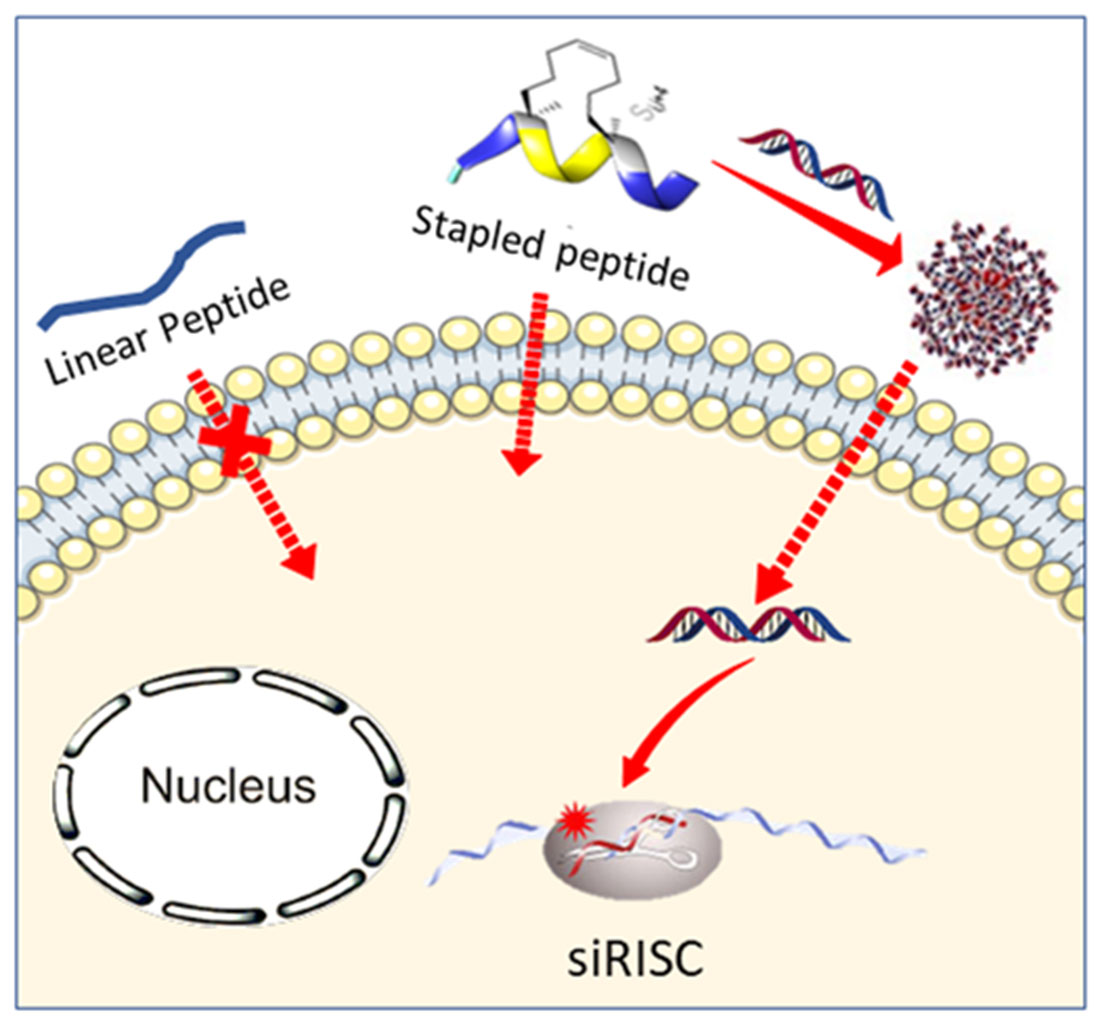
Abstract
Small interfering RNAs (siRNAs) are promising molecules for developing new therapies based on gene silencing; however, their delivery into cells remains an issue. In this study, we took advantage of stapled peptide technology that has emerged as a valuable strategy to render natural peptides more structured, resistant to protease degradation and more bioavailable, to develop short carriers for siRNA delivery. From the pool of stapled peptides that we have designed and synthesized, we identified non-toxic vectors that were able to efficiently encapsulate siRNA, transport them into the cell and induce gene silencing. Remarkably, the most efficient stapled peptide (JMV6582), is composed of only eight amino-acids and contains only two cationic charges.
Nano-assemblies with core-forming hydrophobic polypeptide via polymerization-induced self-assembly (PISA)
Polym. Chem., 2021,12, 113-121 DOI: 10.1002/macp.202000311
Dao T, Vezenkov L, Subra G, Ladmiral V, Semsarilar M.
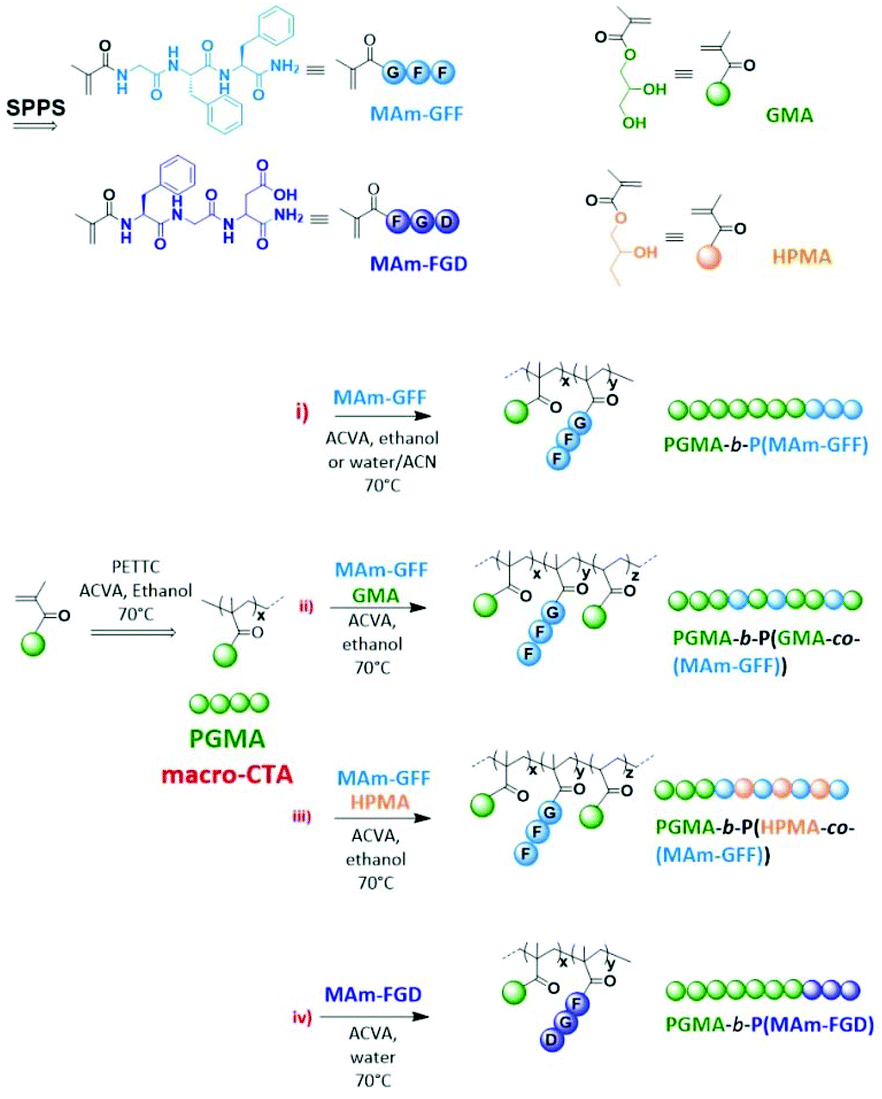
Abstract
The aim of this study is to produce self-assembled structures with hydrophobic polypeptide cores via Reversible Addition–Fragmentation chain Transfer (RAFT) – mediated Polymerisation-Induced Self-Assembly (PISA). Hydrophilic poly(glycerol monomethacrylate) macromolecular chain transfer agents (PGMA mCTAs) were used to polymerize the self-assembling peptide monomers, resulting in the formation of diblock copolymer nano objects. Methacrylamide derivatives containing self-assembling tripeptides MAm-GFF (MAm-Gly-Phe-Phe-NH2) and MAm-FGD (MAm-Phe-Gly-Asp-NH2) were used as hydrophobic monomers. The self-assembling behaviours of these monomers mainly derive from the interactions of the phenylalanine residues, however their difference in hydrophobicity required different polymerization conditions. MAm-GFF was polymerized in the presence of organic solvent (ethanol or acetonitrile), under either dispersion or emulsion polymerization, while MAm-FGD was polymerized under aqueous dispersion conditions. PGMA-b-P(MAm-FGD) obtained from aqueous PISA typically formed fibrous structures while a range of morphologies such as fibre-, flake-, and leaf-like or spherical vesicles were obtained for PGMA-b-P(MAm-GFF) depending on the copolymer composition and solvent used. In all cases the peptides self-assembling core had a crucial influence on the final morphologies.
Bottom-up strategies for the synthesis of peptide-based polymers
Progress in Polymer Science 115 (2021) 101377 https://doi.org/10.1016/j.progpolymsci.2021.101377
Martin J, Desfoux A, Martinez J, Amblard M, Mehdi A, Vezenkov L, Subra G
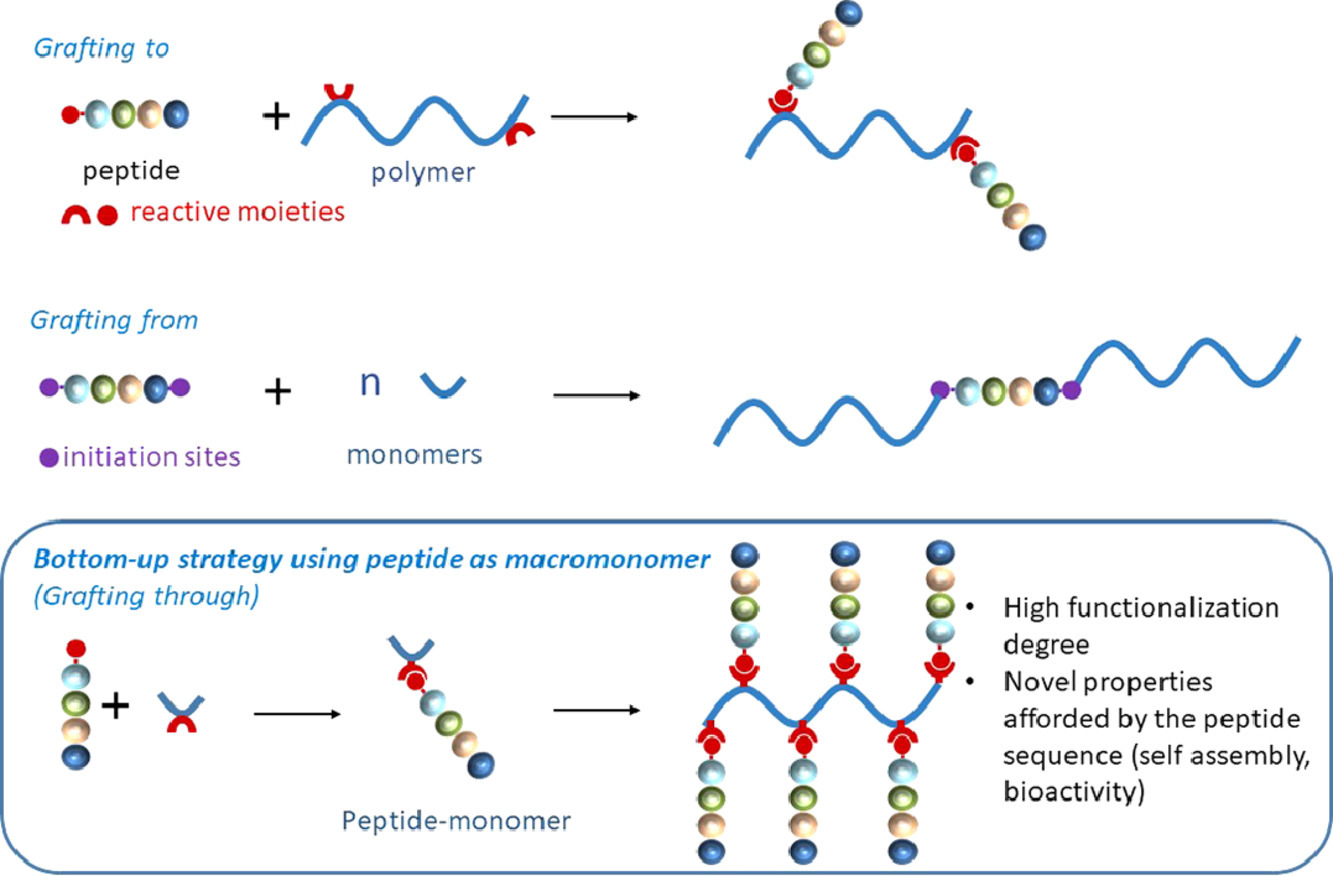
Abstract
Thanks to their wide range of biological activities, peptides have been extensively used to afford designed materials with tailored properties. Peptides can be associated to polymers combining the properties of various polymer backbones with those of bioactive peptide sequences. Such conjugates find promising applications in medical devices, tissue engineering, drug targeting and delivery. Improvement of existing polymers by post-modification peptide grafting is achieved through an extensive range of organic reactions, involving the prior preparation of functional polymers displaying suitable anchoring functions. Alternatively, peptides can be used as initiators of polymerization yielding a chimeric molecule bearing a single peptide at the end of macromolecular chains. Finally, novel polymer materials can be designed when the peptide itself is used as a macromonomer. In that case, the unmatched level of repetition of the peptide sequence or/and its self-assembly properties allow to access very high functionalization degree, original structures and bioactivities.
Self-Assembling Peptide—Polymer Nano-Objects via Polymerization-Induced Self-Assembly
Macromolecules 2020, 53, 16, 7034–7043 https://doi.org/10.1021/acs.macromol.0c01260
Dao T, Vezenkov L, Subra G, Amblard M, In M, Le Meins J-F, Aubrit F, Moradi M-A, Ladmiral A, Semsarilar M*
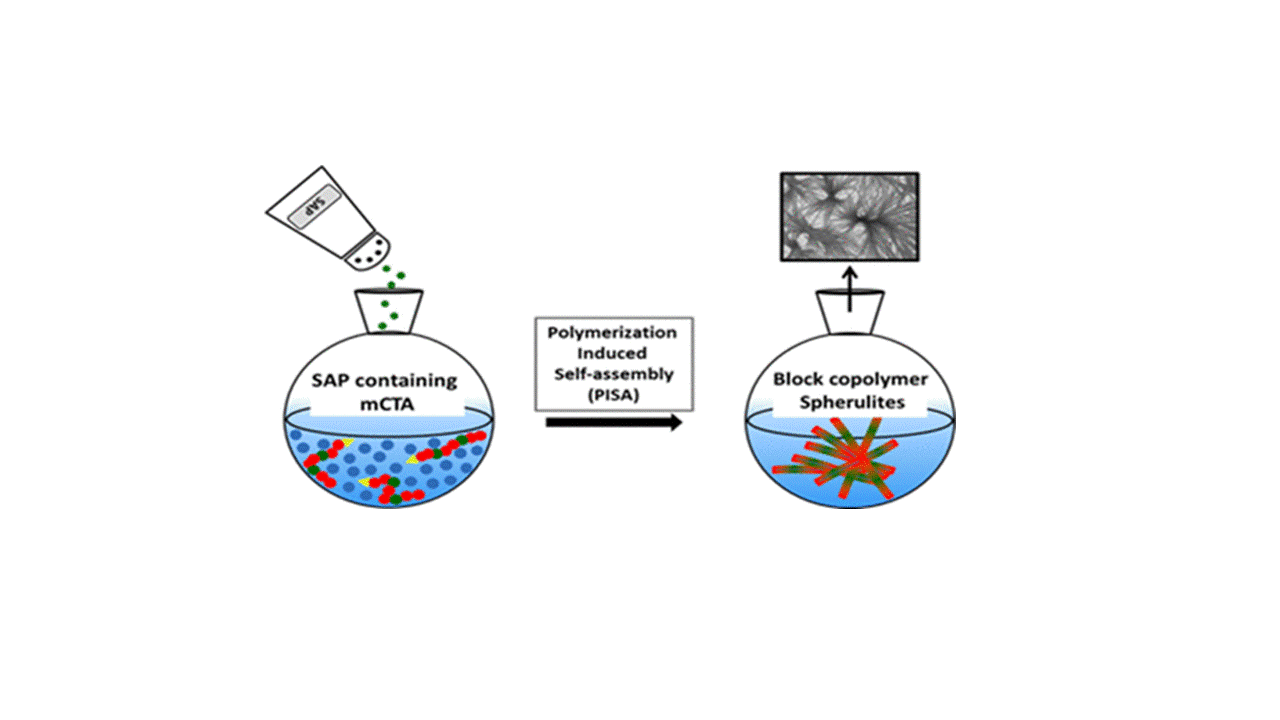
Abstract
Self-assembling peptides (SAPs) have been extensively studied for their ability to form nanoscale ordered structures driven by noncovalent molecular interactions. Meanwhile, polymerization-induced self-assembly (PISA) has been exploited as a facile and efficient way to produce various amphiphilic block copolymer nano-objects, whose self-assembly was governed predominantly by the interactions of the different blocks with the polymerization medium. In this work, we combined PISA with SAPs to prepare novel peptide–polymer hybrid nano-objects, thus harnessing the advantages of PISA and the self-assembling driving force of SAPs. A tripeptide methacrylamide derivative (MAm-Gly-Phe-Phe-NH2, denoted as MAm-GFF, where MAm means methacrylamide) was copolymerized with glycerol monomethacrylate (GMA) to produce a P(GMA65–stat-(MAm-GFF)7) macro-chain transfer agent (macro-CTA) by reversible addition–fragmentation chain transfer polymerization in dimethylformamide. This peptide-based macro-CTA was then successfully chain-extended with poly(2-hydroxypropyl methacrylate) (PHPMA) by aqueous dispersion PISA, forming P(GMA65–stat-(MAm-GFF)7)-b-PHPMA28 self-assembled objects. Fibrous structures were observed by transmission electron microscopy (TEM) and Cryo-TEM, in agreement with depolarized dynamic light scattering, static light scattering, and small-angle X-ray scattering experiments that also revealed long anisotropic morphologies. Such structures have not been reported previously for PISA-prepared nano-objects. This confirms the decisive influence of the GFF SAP on the self-assembly. In addition, annealing the PISA suspension at different temperatures led to a significant size decrease in the self-assembled objects and to a morphological transition caused by the thermosensitivity of both the core-forming PHPMA block and the stabilizing P(GMA-stat-(MAm-GFF)) block.
Evolutionary Divergence of Enzymatic Mechanisms for Tubulin Detyrosination
Cell Rep. 2019 Dec 17;29(12):4159-4171.e6. doi: 10.1016/j.celrep.2019.11.074.
Van der Laan S, Lévêque MF, Marcellin G, Vezenkov L, Lannay Y, Dubra G, Bompard G, Ovejero S, Urbach S, Burgess A, Amblard M, Sterkers Y, Bastien P, Rogowski K.
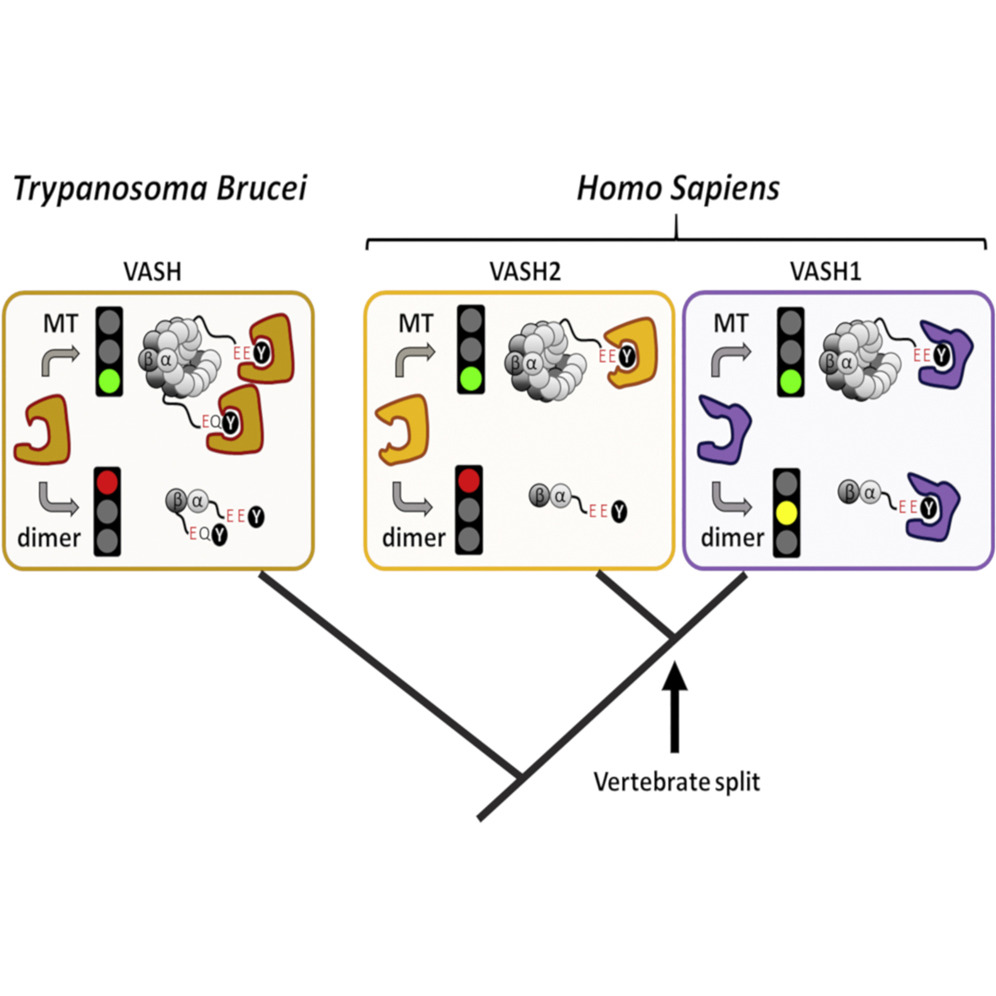
Abstract
The two related members of the vasohibin family, VASH1 and VASH2, encode human tubulin detyrosinases. Here we demonstrate that, in contrast to VASH1, which requires binding of small vasohibin binding protein (SVBP), VASH2 has autonomous tubulin detyrosinating activity. Moreover, we demonstrate that SVBP acts as a bona fide activator of both enzymes. Phylogenetic analysis of the vasohibin family revealed that regulatory diversification of VASH-mediated tubulin detyrosination coincided with early vertebrate evolution. Thus, as a model organism for functional analysis, we used Trypanosoma brucei (Tb), an evolutionarily early-branched eukaryote that possesses a single VASH and encodes a terminal tyrosine on both α- and β-tubulin tails, both subject to removal. Remarkably, although detyrosination levels are high in the flagellum, TbVASH knockout parasites did not present any noticeable flagellar abnormalities. In contrast, we observed reduced proliferation associated with profound morphological and mitotic defects, underscoring the importance of tubulin detyrosination in cell division.
A new quinoline-based chemical probe inhibits the autophagy-related cysteine protease ATG4B
Sci Rep. 2018 Aug 3;8(1):11653. doi: 10.1038/s41598-018-29900-x.
Bosc D, Vezenkov L, Bortnik S, An J, Xu J, Choutka C, Hannigan AM, Kovacic S, Loo S, Clark PGK, Chen G, Guay-Ross RN, Yang K, Dragowska WH, Zhang F, Go NE, Leung A, Honson NS, Pfeifer TA, Gleave M, Bally M, Jones SJ, Gorski SM, Young RN.
Abstract
The cysteine protease ATG4B is a key component of the autophagy machinery, acting to proteolytically prime and recycle its substrate MAP1LC3B. The roles of ATG4B in cancer and other diseases appear to be context dependent but are still not well understood. To help further explore ATG4B functions and potential therapeutic applications, we employed a chemical biology approach to identify ATG4B inhibitors. Here, we describe the discovery of 4-28, a styrylquinoline identified by a combined computational modeling, in silico screening, high content cell-based screening and biochemical assay approach. A structure-activity relationship study led to the development of a more stable and potent compound LV-320. We demonstrated that LV-320 inhibits ATG4B enzymatic activity, blocks autophagic flux in cells, and is stable, non-toxic and active in vivo. These findings suggest that LV-320 will serve as a relevant chemical tool to study the various roles of ATG4B in cancer and other contexts
Ribbon-like Foldamers for Cellular Uptake and Drug Delivery
Chembiochem 2017 Nov 2;18(21):2110-2114. doi: 10.1002/cbic.201700455. Epub 2017 Sep 22.
Vezenkov LL, Martin V, Bettache N, Simon M, Messerschmitt A, Legrand B, Bantignies JL, Subra G, Maynadier M, Bellet V, Garcia M, Martinez J, Amblard M.
Abstract
Different intracellular delivery systems of bioactive compounds have been developed, including cell-penetrating peptides. Although usually nontoxic and biocompatible, these vectors share some of the general drawbacks of peptides, notably low bioavailability and susceptibility to protease degradation, that limit their use. Herein, the conversion of short peptide sequences into poly-α-amino-γ-lactam foldamers that adopt a ribbon-like structure is investigated. This template is used to distribute critical cationic and/or hydrophobic groups on both sides of the backbone, leading to potent short, cell-permeable foldamers with a low positive-charge content. The lead compound showed dramatically improved protease resistance and was able to efficiently deliver a biologically relevant cargo inside cells. This study provided a simple strategy to convert short peptide sequences into efficient protease-resistant cell-penetrating foldamers.
Structure-Activity Relationships of JMV4463, a Vectorized Cathepsin D Inhibitor with Antiproliferative Properties: The Unique Role of the AMPA-Based Vector
ChemMedChem, 2016, Volume: 11, Issue: 3, Pages: 302-308, DOI: 10.1002/cmdc.201500457
L. Vezenkov, C. A. Sanchez, V. Bellet, V. Martin, M. Maynadier, N. Bettache, V. Lisowski, J. Martinez, M. Garcia, M. Amblard, J. F. Hernandez
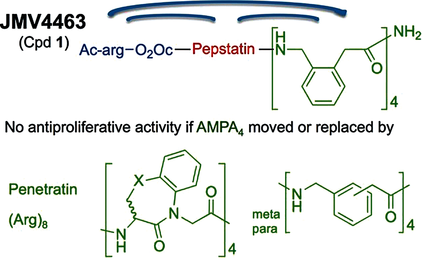
Abstract
Cathepsin D (CathD) is overexpressed and secreted by several solid tumors and stimulates their growth, the mechanism of which is still not understood. In this context, the pepstatin bioconjugate JMV4463 [Ac-arg-O2Oc-(Val)3-Sta-Ala-Sta-(AMPA)4-NH2; O2Oc=8-amino-3,6-dioxaoctanoyl, Sta=statine, AMPA=ortho-aminomethylphenylacetyl], contg. a new kind of cell-penetrating vector, was previously shown to exhibit potent antiproliferative effects in vitro and to delay the onset of tumors in vivo. In this study, the authors performed a structure-activity relationship anal. to evaluate the significance of the inhibitor and vector moieties of JMV4463. By modifying both statine residues of pepstatin the authors found that the antiproliferative activity is correlated with CathD inhibition, supporting a major role of the catalytic activity of intracellular CathD in cancer cell proliferation. Replacing the vector composed of four AMPA units with other vectors was found to abolish cytotoxicity, although all of the conjugates enabled pepstatin transport into cells. In addn., the AMPA4 vector must be localized at the C terminus of the bioconjugate. The unexpected importance of the vector structure and position for cytotoxic action suggests that AMPA4 enables pepstatin to inhibit the proteolysis of crit. CathD substrates involved in cell proliferation via a unique mechanism of action.
Turning Peptide Sequences into Ribbon Foldamers by a Straightforward Multicyclization Reaction
Angewandte Chemie, International Edition, 2015, Volume: 54, Issue: 47, Pages: 13966-13970, DOI: 10.1002/anie.201506955
V. Martin, B. Legrand, L. L. Vezenkov, M. Berthet, G. Subra, M. Calmes, J-L. Bantignies, J. Martinez, M. Amblard

Abstract
The conformational control of mol. scaffolds allows the display of functional groups in defined spatial arrangement. This is of considerable interest for developing fundamental and applied systems in both the fields of biol. and material sciences. Peptides afford a large diversity of functional groups, and peptide synthetic routes are very attractive and accessible. However, most short peptides do not possess well-defined secondary structures. Herein, we developed a simple strategy for converting peptide sequences into structured γ-lactam-contg. oligomers while keeping the amino acids side chain diversity. We showed the propensity of these mols. to adopt ribbon-like secondary structures. The periodic distribution of the functional groups on both sides of the ribbon plane is encoded by the initial peptide sequence.
