
LUC BRUNEL
CNRS EnGIneer
Luc BRUNEL graduated from a DEA in heterocycle chemistry, polymers and catalysis obtained at the University of Montpellier in 1997. After several experiences in the private sector, since 2003 he has been working as a CNRS engineer in biomolecule development.
He works on both the SynBio3 platform providing biomolecules of biological interest and pharmaceuticals, as responsible for syntheses and purifications in large quantities, but also within the research team of the laboratory to carry out certain precursor projects.
.Luc has very solid skills in the fields of peptide synthesis in solution and on solid support, with a specialty in the field of sulfo and phosphopeptides.
.He also has 20 years of expertise in the implementation, development of new methods and scale-up in purification by preparative HPLC.
Luc obtained the CNRS Crystal medal in 2015.
Contact:
luc.brunel@umontpellier.fr
+33 (0)4 48 79 22 37
5 major publications :
A Collagen-Mimetic Organic-Inorganic Hydrogel for Cartilage Engineering. Valot L, Maumus M, Brunel L, Martinez J, Amblard M, Noël D, Mehdi A, Subra G. Gels. 2021 Jun 15;7(2):73. doi: 10.3390/gels7020073
In Vivo Stabilization of a Gastrin-Releasing Peptide Receptor Antagonist Enhances PET Imaging and Radionuclide Therapy of Prostate Cancer in Preclinical Studies. Chatalic KL, Konijnenberg M, Nonnekens J, de Blois E, Hoeben S, de Ridder C, Brunel L, Fehrentz JA, Martinez J, van Gent DC, Nock BA, Maina T, van Weerden WM, de Jong M. Theranostics. 2016 Jan 1;6(1):104-17. doi: 10.7150/thno.13580. eCollection 2016.
Gastrin releasing peptide receptor-directed radioligands based on a bombesin antagonist: synthesis, (111)in-labeling, and preclinical profile. Marsouvanidis PJ, Nock BA, Hajjaj B, Fehrentz JA, Brunel L, M’Kadmi C, van der Graaf L, Krenning EP, Maina T, Martinez J, de Jong M. J Med Chem. 2013 Mar 28;56(6):2374-84. doi: 10.1021/jm301692p. Epub 2013 Mar 8.
An innovative strategy for sulfopeptides analysis using MALDI-TOF MS reflectron positive ion mode. Cantel S, Brunel L, Ohara K, Enjalbal C, Martinez J, Vasseur JJ, Smietana M. Proteomics. 2012 Aug;12(14):2247-57. doi: 10.1002/pmic.201100525.
The 1,2,4-triazole as a scaffold for the design of ghrelin receptor ligands: development of JMV 2959, a potent antagonist. Moulin A, Brunel L, Boeglin D, Demange L, Ryan J, M’Kadmi C, Denoyelle S, Martinez J, Fehrentz JA. Amino Acids. 2013 Feb;44(2):301-14. doi: 10.1007/s00726-012-1355-2. Epub 2012 Jul 14. Review.

Involvement of PPARγ in the Anticonvulsant Activity of EP-80317, a Ghrelin Receptor Antagonist
Front Pharmacol. 2017 Sep 22;8:676. doi: 10.3389/fphar.2017.00676. eCollection 2017.
Lucchi C, Costa AM, Giordano C, Curia G, Piat M, Leo G, Vinet J, Brunel L, Fehrentz JA, Martinez J, Torsello A, Biagini G.
Abstract
Ghrelin, des-acyl ghrelin and other related peptides possess anticonvulsant activities. Although ghrelin and cognate peptides were shown to physiologically regulate only the ghrelin receptor, some of them were pharmacologically proved to activate the peroxisome proliferator-activated receptor gamma (PPARγ) through stimulation of the scavenger receptor CD36 in macrophages. In our study, we challenged the hypothesis that PPARγ could be involved in the anticonvulsant effects of EP-80317, a ghrelin receptor antagonist. For this purpose, we used the PPARγ antagonist GW9662 to evaluate the modulation of EP-80317 anticonvulsant properties in two different models. Firstly, the anticonvulsant effects of EP-80317 were studied in rats treated with pilocarpine to induce status epilepticus (SE). Secondly, the anticonvulsant activity of EP-80317 was ascertained in the repeated 6-Hz corneal stimulation model in mice. Behavioral and video electrocorticographic (ECoG) analyses were performed in both models. We also characterized levels of immunoreactivity for PPARγ in the hippocampus of 6-Hz corneally stimulated mice. EP-80317 predictably antagonized seizures in both models. Pretreatment with GW9662 counteracted almost all EP-80317 effects both in mice and rats. Only the effects of EP-80317 on power spectra of ECoGs recorded during repeated 6-Hz corneal stimulation were practically unaffected by GW9662 administration. Moreover, GW9662 alone produced a decrease in the latency of tonic-clonic seizures and accelerated the onset of SE in rats. Finally, in the hippocampus of mice treated with EP-80317 we found increased levels of PPARγ immunoreactivity. Overall, these results support the hypothesis that PPARγ is able to modulate seizures and mediates the anticonvulsant effects of EP-80317.
Sol-gel synthesis of collagen-inspired peptide hydrogel
Materials Today , Pages: Ahead of Print, Journal, 2017, DOI: 10.1016/j.mattod.2017.02.001
C. Echalier, S. Jebors, G. Laconde, B. Guillaume, V. Luc, C. Pascal, B. Lea, A. Bethry, B. Legrand, H. Van Den Berghe, X. Garric, D. Noel, J. Martinez, A. Mehdi, G. Subra
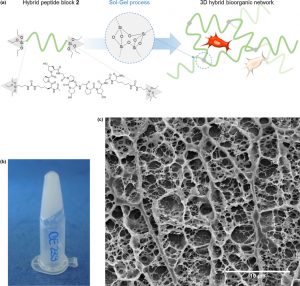
Abstract
Conceiving biomaterials able to mimic the specific environments of extracellular matrixes are a prerequisite for tissue engineering applications. Numerous types of polymers (PEG, PLA, etc.) have been used for the design of biocompatible scaffolds, but they are still less efficient than natural biopolymers such as collagen exts. Chem. modified and loaded with different bioactive factors, biopolymers afford an environment favorable to cell proliferation and differentiation. Unfortunately, they present several drawbacks, such as weak batch-to-batch reproducibility, potential immunogenicity and high cost of prodn. Herein we propose a fully synthetic covalent hydrogel obtained by sol-gel polymn. of a silylated peptide. We selected a short and low mol. building-block derived from the consensus collagen sequence [Pro-Hyp-Gly]. Interestingly, the sol-gel process occurs in physiol. buffer, enabling the embedment of stem cells. This collagen-inspired hydrogel provides a cell-friendly environment comparable to natural collagen substrates, demonstrating its potency as a biomimetic scaffold.
In vivo stabilization of a gastrin-releasing peptide receptor antagonist enhances PET imaging and radionuclide therapy of prostate cancer in preclinical studies
Theranostics, 2016, Volume: 6, Issue: 1, Pages: 104-117, DOI: 10.7150/thno.13580
K. L. S. Chatalic, M. Konijnenberg, J. Nonnekens, H. de Blois, S. Erik, C. de Ridder, L. Brunel, J. A. Fehrentz, J. Martinez, D. C. van Gent, B. A. Nock, T. Maina, W. M. van Weerden, M. de Jong
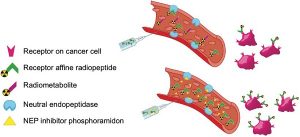
Abstract
A single tool for early detection, accurate staging, and personalized treatment of prostate cancer (PCa) would be a major breakthrough in the field of PCa. Gastrin-releasing peptide receptor (GRPR) targeting peptides are promising probes for a theranostic approach for PCa overexpressing GRPR. However, the successful application of small peptides in a theranostic approach is often hampered by their fast in vivo degrdn. by proteolytic enzymes, such as neutral endopeptidase (NEP). Here we show for the first time that co-injection of a NEP inhibitor (phosphoramidon (PA)) can lead to an impressive enhancement of diagnostic sensitivity and therapeutic efficacy of the theranostic 68Ga-/177Lu-JMV4168 GRPR-antagonist. Co-injection of PA (300 μg) led to stabilization of 177Lu-JMV4168 in murine peripheral blood. In PC-3 tumor-bearing mice, PA co-injection led to a two-fold increase in tumor uptake of 68Ga-/177Lu-JMV4168, 1 h after injection. In positron emission tomog. (PET) imaging with 68Ga-JMV4168, PA co-injection substantially enhanced PC-3 tumor signal intensity. Radionuclide therapy with 177Lu-JMV4168 resulted in significant regression of PC-3 tumor size. Radionuclide therapy efficacy was confirmed by prodn. of DNA double strand breaks, decreased cell proliferation and increased apoptosis. Increased survival rates were obsd. in mice treated with 177Lu-JMV4168 plus PA as compared to those without PA. This data shows that co-injection of the enzyme inhibitor PA greatly enhances the theranostic potential of GRPR-radioantagonists for future application in PCa patients.
In vitro and in vivo application of radiolabeled gastrin-releasing peptide receptor ligands in breast cance
Journal of Nuclear Medicine, 2015, Volume: 56, Issue: 5, Pages: 752-757, DOI: 10.2967/jnumed.114.153023
S. U. Dalm, J. W. M. Martens, A. M. Sieuwerts, C. H. M. van Deurzen, S. J. Koelewijn, E. de Blois, T. Maina, B. A. Nock, L. Brunel, J.-A. Fehrentz, J. Martinez, M. de Jong, M. Melis
Abstract
Breast cancer (BC) consists of multiple subtypes defined by various mol. characteristics, for instance, estrogen receptor (ER) expression. Methods for visualizing BC include mammog., MR imaging, ultrasound, and nuclear medicine-based methods such as 99mTc-sestamibi and 18F-FDG PET, unfortunately all lacking specificity. Peptide receptor scintigraphy and peptide receptor radionuclide therapy are successfully applied for imaging and therapy of somatostatin receptor-expressing neuroendocrine tumors using somatostatin receptor radioligands. On the basis of a similar rationale, radioligands targeting the gastrin-releasing peptide receptor (GRP-R) might offer a specific method for imaging and therapy of BC. The aim of this study was to explore the application of GRP-R radioligands for imaging and therapy of BC by introducing valid preclin. in vitro and in vivo models. Methods: GRP-R expression of 50 clin. BC specimens and the correlation with ER expression was studied by in vitro autoradiog. with the GRP-R agonist 111In-AMBA. GRP-R expression was also analyzed in 9 BC cell lines applying 111In-AMBA internalization assays and quant. reverse transcriptase polymerase chain reaction. In vitro cytotoxicity of 111Lu-AMBA was detd. on the GRP-R-expressing BC cell line T47D. SPECT/CT imaging and biodistribution were studied in mice with s.c. and orthotopic ER-pos. T47D and MCF7 xenografts after injection of the GRP-R antagonist 111In-JMV4168. Results: Most of the human BC specimens (96%) and BC cell lines (6/9) were found to express GRP-R. GRP-R tumor expression was pos. (P = 0.026, Χ2(4) = 12,911) correlated with ER expression in the human BC specimens. Treatment of T47D cells with 10-7 M/50 MBq of 111Lu-AMBA resulted in 80% redn. of cells in vitro. Furthermore, s.c. and orthotopic tumors from both BC cell lines were successfully visualized in vivo by SPECT/CT using 111In-JMV4168; T47D tumors exhibited a higher uptake than MCF7 xenografts. Conclusion: Targeting GRP-R-expressing BC tumors using GRP-R radioligands is promising for nuclear imaging and therapy, esp. in ER-pos. BC patients.
Preclinical comparison of Al18F- and 68Ga-labeled gastrin-releasing peptide receptor antagonists for PET imaging of prostate cancer
Journal of Nuclear Medicine, 2014, Volume: 55, Issue: 12, Pages: 2050-2056, DOI: 10.2967/jnumed.114.141143
K. l. L. S. Chatalic, G. M. Franssen, W. M. van Weerden, W. J. McBride, P. Laverman, E. de Blois, B. Hajjaj, L. Brunel, D. M. Goldenberg, J.-A. Fehrentz, J. Martinez, O. C. Boerman, M. de Jong,
Abstract
Gastrin-releasing peptide receptor (GRPR) is overexpressed in human prostate cancer and is being used as a target for mol. imaging. In this study, we report on the direct comparison of 3 novel GRPR-targeted radiolabeled tracers: Al18F-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 (JMV5132 is NODA-MPAA-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], JMV4168 is DOTA-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], and NODA-MPAA is 2-[4-(carboxymethyl)-7-{[4-(carboxymethyl) phenyl]methyl}-1,4,7-triazacyclononan-1-yl]acetic acid). Methods: The GRPR antagonist JMV594 (H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) was conjugated to NODA-MPAA for labeling with Al18F. JMV5132 was radiolabeled with 68Ga and 18F, and JMV4168 was labeled with 68Ga for comparison. The inhibitory concn. of 50% values for binding GRPR of JMV4168, JMV5132, natGa-JMV4168, and natGa-JMV5132 were detd. in a competition-binding assay using GRPR-overexpressing PC-3 tumors. The tumor-targeting characteristics of the compds. were assessed in mice bearing s.c. PC-3 xenografts. Small-animal PET/CT images were acquired, and tracer biodistribution was detd. by ex vivo measurements. Results: JMV5132 was labeled with 18F in a novel 1-pot, 1-step procedure within 20 min, without need for further purifn. and resulting in a specific activity of 35 MBq/nmol. Inhibitory concn. of 50% values (in nM) for GRPR binding of JMV5132, JMV4168, natGa-JMV5132, natGa-JMV4168, and AlnatF-JMV5132 were 6.8 (95% confidence intervals [CIs], 4.6-10.0), 13.2 (95% CIs, 5.9-29.3), 3.0 (95% CIs, 1.5-6.0), 3.2 (95% CIs, 1.8-5.9), and 10.0 (95% CIs, 6.3-16.0), resp. In mice with s.c. PC-3 xenografts, all tracers cleared rapidly from the blood, exclusively via the kidneys for 68Ga-JMV4168 and partially hepatobiliary for 68Ga-JMV5132 and Al18F-JMV5132. Two hours after injection, the uptake of 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 in PC-3 tumors was 5.96 ± 1.39, 5.24 ± 0.29, 5.30 ± 0.98 (percentage injected dose per g), resp. GRPR specificity was confirmed by significantly reduced tumor uptake of the 3 tracers after coinjection of a 100-fold excess of unlabeled JMV4168 or JMV5132. Small-animal PET/CT clearly visualized PC-3 tumors, with the highest resoln. obsd. for Al18F-JMV5132. Conclusion: JMV5132 could be rapidly and efficiently labeled with 18F. Al18F-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 all showed comparable high and specific accumulation in GRPR-pos. PC-3 tumors. These new PET tracers are promising candidates for future clin. translation.
Ghrelin Receptor Ligands: Design and Synthesis of Pseudopeptides and Peptidomimetics
Current Chemical Biology, 2013, Volume: 7, Issue: 3, Pages: 254-270, , DOI: 10.2174/2212796807999131128125920
A. Moulin, L. Brunel, P. Verdie, L. Gavara, J. Martinez, J-A. Fehrentz
Abstract
A review. Mainly synthesized in the stomach, ghrelin is a peptide hormone which stimulates growth hormone secretion and appetite, thus promoting food intake and body-wt. gain. Historically, researchers started to work on the discovery of ghrelin receptor ligands several years before the discovery of the ghrelin receptor and the hormone itself. Indeed peptides able to stimulate growth hormone secretion (growth hormone releasing peptides, GHRPs) were found while the mechanism of action and the target receptor were still unknown. Non peptidic agonists were then described (growth hormone secretagogues, GHSs) and the receptor (GHS-R1a) identified in 1996. Three years later, the natural ligand of this receptor (ghrelin) was isolated from stomach and its chem. synthesis allowed to show the physiol. role of ghrelin in energy balance. In this review, we present some pseudopeptide and peptidomimetic approaches used by researchers for the design of ghrelin receptor ligands. We will start by the pioneering work of Bowers et al. on enkephalin analogs, which was the starting point for the development of an impressive no. of compds., by several of the major worldwide pharma companies. We will also describe the work achieved starting from a substance P deriv., which was one of the first peptides identified as an antagonist of the newly discovered ghrelin receptor. Then we will review the structure activity relationship study starting from the peptide ghrelin, which started with the discovery of this peptide in 1999. We will also focus on a more recent work based on macrocyclic peptidic analogs for the development of ghrelin receptor ligands.
The 1, 2, 4-triazole as a scaffold for the design of ghrelin receptor ligands: development of JMV 2959, a potent antagonist
Amino Acids. 2013 Feb;44(2):301-14. doi: 10.1007/s00726-012-1355-2. Epub 2012 Jul 14. Review.
Moulin A, Brunel L, Boeglin D, Demange L, Ryan J, M’Kadmi C, Denoyelle S, Martinez J, Fehrentz JA.
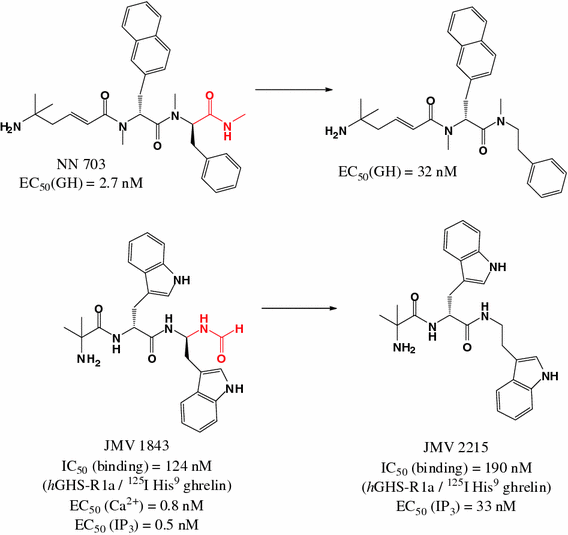
Abstract
Ghrelin is a 28-residue peptide acylated with an n-octanoyl group on the Ser 3 residue, predominantly produced by the stomach. Ghrelin displays strong growth hormone (GH) releasing activity, which is mediated by the activation of the so-called GH secretagogue receptor type 1a (GHS-R1a). Given the wide spectrum of biological activities of Ghrelin in neuroendocrine and metabolic pathways, many research groups, including our group, developed synthetic peptide, and nonpeptide GHS-R1a ligands, acting as agonists, partial agonists, antagonists, or inverse agonists. In this highlight article, we will focus on the discovery of a GHS-R1a antagonist compound, JMV 2959, which has been extensively studied in different in vitro and in vivo models. We will first describe the peptidomimetic approach that led us to discover this compound. Then we will review the results obtained with this compound in different studies in the fields of food intake and obesity, addictive behaviors, hyperactivity and retinopathy.
