ACS Omega. 2019 May 22;4(5):8862-8873. doi: 10.1021/acsomega.9b00465. eCollection 2019 May 31
Maillard LT, Park HS, Kang YK.
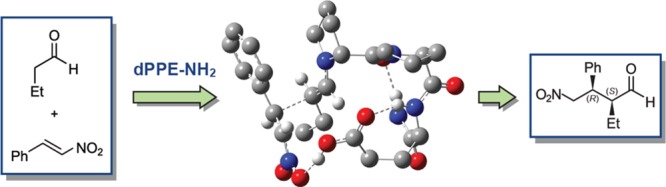
Abstract
The mechanism of the asymmetric addition of aldehyde (butanal) to nitroolefin (β-nitrostyrene) catalyzed by H-d-Pro-Pro-Glu-NH2 (dPPE-NH2; 1) was explored using density functional theory methods in chloroform. By conformational search, it was confirmed that catalyst 1 and its enamine intermediate adopted a dominant conformation with a βI structure stabilized by a C10 H-bond between the C=O of d-Pro1 and C-terminal NH2 proton and by an additional H-bond between the side chain and the backbone of Glu3. This βI turn structure was conserved all along the catalytic cycle. Consistently with the kinetic studies, the C-C bond formation between the enamine and electrophile was also confirmed as the rate-determining step. The stereoselectivity results from a re → re prochiral approach of enamine and β-nitrostyrene with a gauche– orientation of the double bonds. Although it was suggested as the possible formation of dihydrooxazine oxide species, this process was confirmed to be kinetically less accessible than the formation of acyclic nitronate. In particular, our calculated results supported that the carboxylic acid group of Glu3 in 1 played a central role by acting as general acid/base all along the catalytic cycle and orienting the asymmetric C-C bond formation.
