
Ludovic Maillard
Assistant Professor, Faculty of Pharmacy, Montpellier University
Ludovic Maillard is Pharmacyst (Paris XI, 2002) and Chemical Engineer (ENSCP, Paris VI, 2002). After a Master Degree in organic chemistry (Paris VI, 2002), he did a PhD in bioorganic chemistry at the ICSN, in the group of Dr. B. Badet. Thereafter he joined the group of Prof. J. Robinson (OCI, Zurich, Switzerland) for a one-year post-doctoral fellow. He is actually Assistant Professor in medicinal chemistry.
One of his major research interests is to develop and characterized conformationaly predictable molecular architectures named foldamers, which are constructed from heterocyclic γ-amino acids. In such a context, he has reported a short chemical route to access orthogonally protected thiazole-based γ-amino acids, which were used as building blocks for designing helical γ-peptide foldmers and antimicrobial peptides. These platforms are highly versatile and efforts are made to expand their applications toward cellular targeting and organo-catalysis.
Contact:
ludovic.maillard@umontpellier.Fr
+33 (0)4 48 79 21 84
5 major publications :
C. Bonnel, B. Legrand, M. Simon, J. Martinez, J.L. Bantignies, Y.K. Kang, E. Wenger, Francois Hoh, N. Masurier, L. T. Maillard* (2017)C9/12 Ribbon-Like Structures in Hybrid Peptides Alternating α- and Thiazole-Based γ-Amino Acids, Chem. Eur. J., 17584-17591
Bonnel C., Legrand B., Bantignies J.-L., Petitjean H., Martinez J., Masurier N. and Maillard L. T.* (2016) FT-IR and NMR structural markers for thiazole-based γ-peptide foldamers Org. Biomol. Chem. 8664-8669.
Mathieu L., Bonnel C., Masurier N., Maillard L. T.*, Martinez J. and Lisowski V. (2015) Cross-Claisen Condensation of N-Fmoc-Amino Acids – A Short Route to Heterocyclic γ-Amino Acids Eur. J. Org. Chem. 2262–2270.
Legrand B., Mathieu L., Lebrun A., Andriamanarivo S., Lisowski V., Masurier N., Zirah S., Kang Y. K., Martinez J., Maillard L.T.*, (2014) Thiazole-Based γ-Building Blocks as Reverse-Turn Mimetic to Design a Gramicidin S Analogue: Conformational and Biological Evaluation Chem. Eur. J. 6713-6720.
Mathieu L., Legrand B., Deng C., Vezenkov L., Wenger E., Didierjean C., Amblard M., Averlant-Petit MC., Masurier N., Lisowski V., Martinez J. and Maillard L.T.* (2013) Helical Oligomers of Thiazole-Based γ-Amino Acids: Synthesis and Structural Studies. Angew. Chem. Int. Ed. 52, 6006-6010.

1,3-Diazepine Derivatives: Strategies for Synthesis
Eur. J. Org. Chem., 2022, 21, e202100492, https://doi.org/10.1002/ejoc.202100492
Y. Malki, L.T. Maillard, N. Masurier
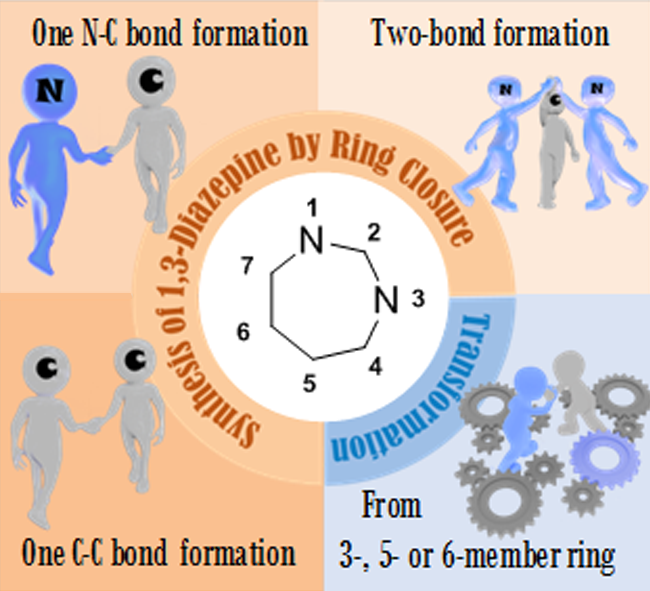
Abstract
The present review describes the methodologies reported for the synthesis of 1,3-diazepine derivatives. This heterocyclic moiety forms the core structure of many compounds with potent biological activities. Synthetic strategies by ring closure or ring transformation are discussed.
α,β-Unsaturated γ-Peptide Foldamers
Chempluschem. 2021 Apr 1;86(4):629-645. doi: 10.1002/cplu.202100045. Online ahead of print.
Legrand B, Maillard LT
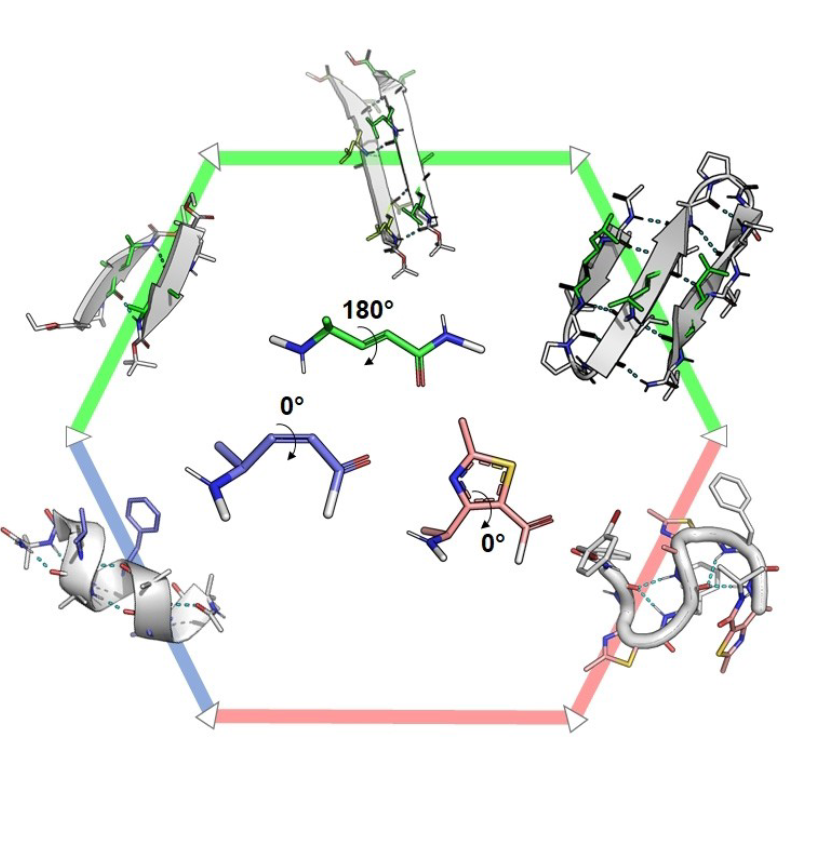
Abstract
Despite their concomitant emergence in the 1990s, γ-peptide foldamers have not developed as fast as β-peptide foldamers and to date, only a few γ-oligomer structures have been reported, and with sparse applications. Among these examples, sequences containing α,β-unsaturated γ-amino acids have recently drawn attention since the Z/E configurations of the double bond provide opposite planar restrictions leading to divergent conformational behaviors, from helix to extended structures. In this Review, we give a comprehensive overview of the developments of γ-peptide foldamers containing α,β-unsaturated γ-amino acids with examples of applications for health and catalysis, as well as materials science.
Tailoring the Physicochemical Properties of Antimicrobial Peptides onto a Thiazole-Based γ-Peptide Foldamer
J. Med. Chem. 2020, 63, 17, 9168–9180. https://doi.org/10.1021/acs.jmedchem.0c00077
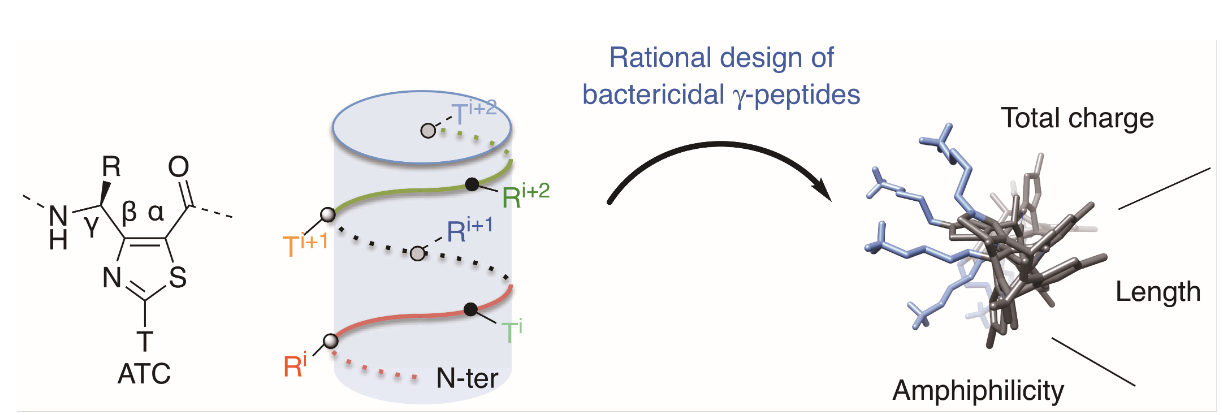
Abstract
Antimicrobial peptides (AMPs) are amphipathic molecules displaying broad-spectrum bactericidal activity, providing opportunities to develop a new generation of antibiotics. However, their use is limited either by poor metabolic stability or by high hemolytic activity. We herein addressed the potential of thiazole-based γ-peptide oligomers named ATCs as tunable scaffolds to design polycationic AMP mimetics. Knowing the side chain distribution along the backbone, we rationally designed facially amphiphilic sequences with bactericidal effect in the micromolar range. Since no hemolytic activity was detected up to 100 μM, this class of compounds has shown the potential for therapeutic development.
Helical γ-peptide foldamers as dual inhibitors of amyloid-β peptide and islet amyloid polypeptide oligomerization and fibrillization
Chem. Eur. J. 26, 14612-14622, https://doi.org/10.1002/chem.202001716
J. Kaffy J., Berardet C., Mathieu L., Legrand B., Taverna M., Halgand F., Van Der Rest G., Maillard L. T., Ongeri S.
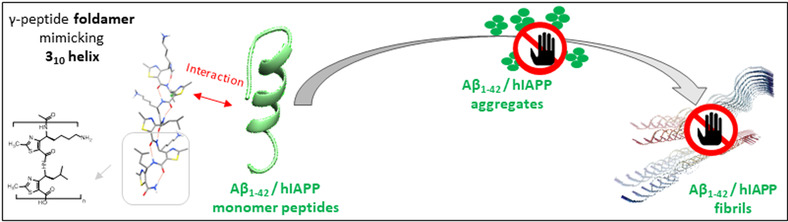
Abstract
Type 2 diabetes (T2D) and Alzheimer’s disease (AD) belong to the 10 deadliest diseases and are sorely lacking in effective treatments. Both pathologies are part of the degenerative disorders named amyloidoses, which involve the misfolding and the aggregation of amyloid peptides, hIAPP for T2D and Aβ1‐42 for AD. While hIAPP and Aβ1‐42 inhibitors have been essentially designed to target β‐sheet‐rich structures composing the toxic amyloid oligomers and fibrils of these peptides, the strategy aiming at trapping the non‐toxic monomers in their helical native conformation has been rarely explored. We report herein the first example of helical foldamers as dual inhibitors of hIAPP and Aβ1‐42 aggregation and able to preserve the monomeric species of both amyloid peptides. A foldamer composed of 4‐amino(methyl)‐1,3‐thiazole‐5‐carboxylic acid (ATC) units, adopting a 9‐helix structure reminiscent of 310 helix, was remarkable as demonstrated by biophysical assays combining thioflavin‐T fluorescence, transmission electronic microscopy, capillary electrophoresis and mass spectrometry.
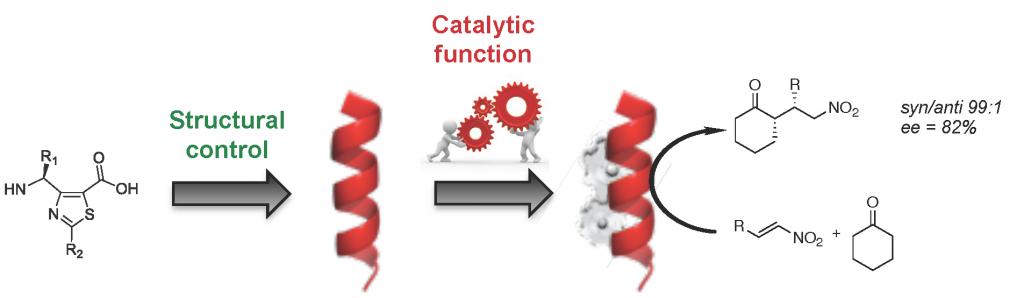
Catalytic Foldamers: When the Structure Guides the Function
Abstract
Enzymes are predominantly proteins able to effectively and selectively catalyze highly complex biochemical reactions in mild reaction conditions. Nevertheless, they are limited to the arsenal of reactions that have emerged during natural evolution in compliance with their intrinsic nature, three-dimensional structures and dynamics. They optimally work in physiological conditions for a limited range of reactions, and thus exhibit a low tolerance for solvent and temperature conditions. The de novo design of synthetic highly stable enzymes able to catalyze a broad range of chemical reactions in variable conditions is a great challenge, which requires the development of programmable and finely tunable artificial tools. Interestingly, over the last two decades, chemists developed protein secondary structure mimics to achieve some desirable features of proteins, which are able to interfere with the biological processes. Such non-natural oligomers, so called foldamers, can adopt highly stable and predictable architectures and have extensively demonstrated their attractiveness for widespread applications in fields from biomedical to material science. Foldamer science was more recently considered to provide original solutions to the de novo design of artificial enzymes. This review covers recent developments related to peptidomimetic foldamers with catalytic properties and the principles that have guided their design.
Organocatalytic Asymmetric Addition of Aldehyde to Nitroolefin by H-d-Pro-Pro-Glu-NH2: A Mechanistic Study
ACS Omega. 2019 May 22;4(5):8862-8873. doi: 10.1021/acsomega.9b00465. eCollection 2019 May 31
Maillard LT, Park HS, Kang YK.
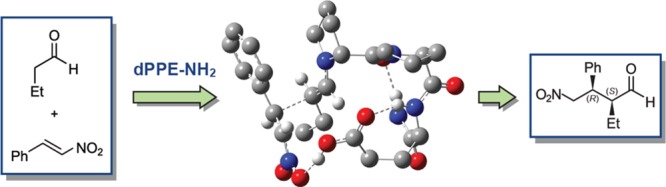
Abstract
The mechanism of the asymmetric addition of aldehyde (butanal) to nitroolefin (β-nitrostyrene) catalyzed by H-d-Pro-Pro-Glu-NH2 (dPPE-NH2; 1) was explored using density functional theory methods in chloroform. By conformational search, it was confirmed that catalyst 1 and its enamine intermediate adopted a dominant conformation with a βI structure stabilized by a C10 H-bond between the C=O of d-Pro1 and C-terminal NH2 proton and by an additional H-bond between the side chain and the backbone of Glu3. This βI turn structure was conserved all along the catalytic cycle. Consistently with the kinetic studies, the C-C bond formation between the enamine and electrophile was also confirmed as the rate-determining step. The stereoselectivity results from a re → re prochiral approach of enamine and β-nitrostyrene with a gauche– orientation of the double bonds. Although it was suggested as the possible formation of dihydrooxazine oxide species, this process was confirmed to be kinetically less accessible than the formation of acyclic nitronate. In particular, our calculated results supported that the carboxylic acid group of Glu3 in 1 played a central role by acting as general acid/base all along the catalytic cycle and orienting the asymmetric C-C bond formation.
Topological Requirements for CI-M6PR-Mediated Cell Uptake
Bioconjugate Chemistry 2019 30 (10), 2533-2538 DOI: 10.1021/acs.bioconjchem.9b00590
Simon M*, Ali LMA*, El Cheikh K, Aguesseau-Kondrotas J, Godefroy A, Nguyen C, Garcia M, Morère A, Gary-Bobo M, Maillard L.
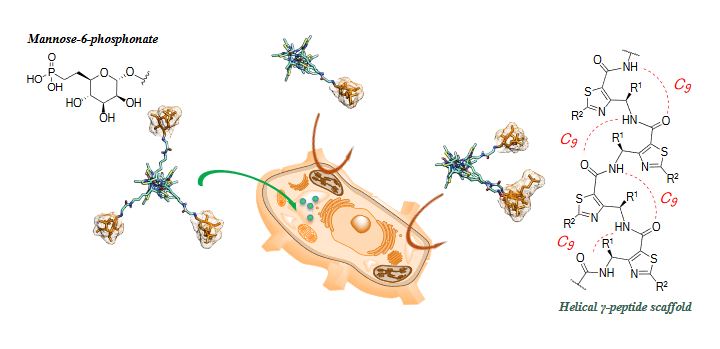
Abstract
The 300 kDa cation-independent M6P receptor (CI-MPR) mediates ligand internalization and trafficking to the endolysosomal compartments. Because of its endocytotic nature, it has been recognized as a promising class of receptors for target component delivery. Its cellular uptake involves the simultaneous binding of two protein units resulting in the formation of receptor dimers. While many multivalent glycoconjugates have been reported to date, little is known about the topological requests to induce an effective recruitment of CI-MPRs. We herein describe the synthesis and cell uptake ability of a set of highly organized glycoclusters bearing one to three saccharide units. The spatial arrangement of carbohydrate ligands is ensured by a heterocyclic γ-peptide central core.
Prospect of Thiazole-based gamma-Peptides Foldamers in Enamine Catalysis: Exploration of the Nitro-Michael Addition
Chemistry. 2019 May 28;25(30):7396-7401. doi: 10.1002/chem.201901221. Epub 2019 May 7.
Aguesseau-Kondrotas J, Simon M, Legrand B, Bantigniès JL, Kang YK, Dumitrescu D, Van der Lee A, Campagne JM, de Figueiredo RM, Maillard LT.
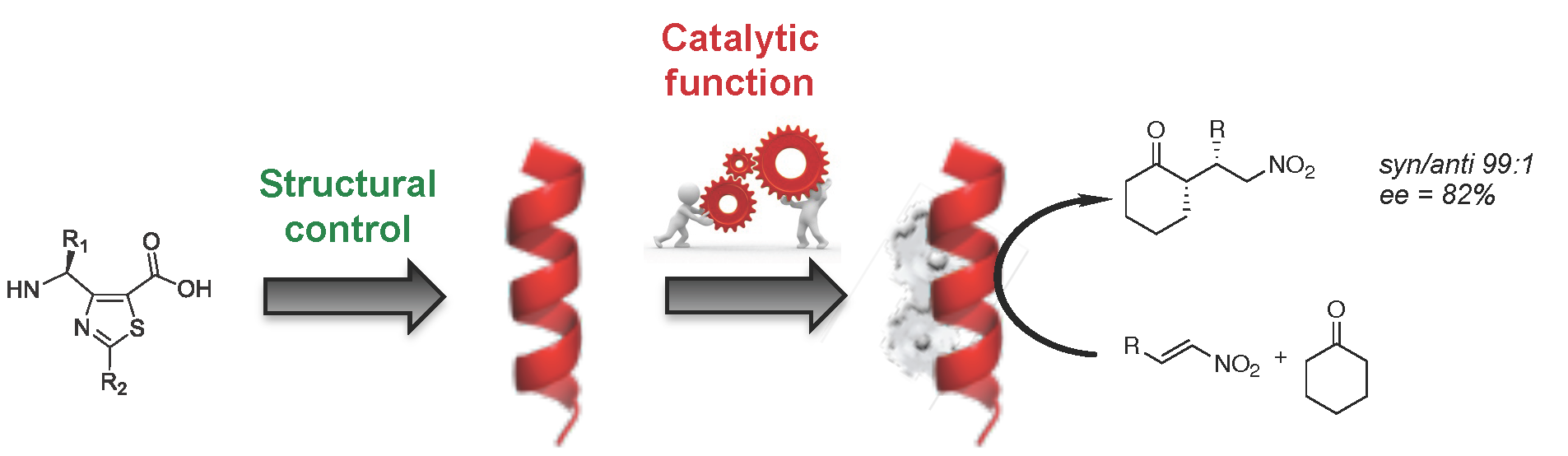
Abstract
As three-dimensional folding is prerequisite to biopolymer activity, complex functions may also be achieved through foldamer science. Because of the diversity of sizes, shapes and folding available with synthetic monomers, foldamer frameworks enable a numerous opportunities for designing new generations of catalysts. We herein demonstrate that heterocyclic γ-peptide scaffolds represent a versatile platform for enamine catalysis. One central feature was to determine how the catalytic activity and the transfer of chiral information might be under the control of the conformational behaviours of the oligomer.
Synthesis of Peptide–Adenine Conjugates as a New Tool for Monitoring Protease Activity
Eur. J. Org. Chem. January 2019: 176-183. doi:10.1002/ejoc.201801490.
Masurier, N. , Soualmia, F. , Sanchez, P. , Lefort, V. , Roué, M. , Maillard, L. T., Subra, G. , Percot, A. and El Amri, C.

Abstract
We took advantage of the powerful adenine SERS (Surface Enhanced Raman Spectroscopy) probe to design peptide–adenine conjugates as candidates for use as serine protease substrates. Whereas the direct introduction of the peptide sequence on the adenine exocyclic N6 amine gave an imidazopurinone derivative, the introduction of an aminoethyl linker between the adenine group and the peptide chain led to the expected candidate probes. These potential substrates were then evaluated for monitoring the hydrolytic activity of trypsin, used as a model protease, by HPLC and by SERS. We demonstrated that the Boc–VPR–adenine conjugate is a substrate of trypsin and constitutes a good starting point to design optimized substrates to monitor protease activity by SERS.
Can Heterocyclic γ-Peptides Provide Polyfunctional Platforms for Synthetic Glycocluster Construction?
Chemistry. 2018 Aug 6;24(44):11426-11432. doi: 10.1002/chem.201802032
Simon M, Ali LMA, El Cheikh K, Aguesseau J, Gary-Bobo M, Garcia M, Morère A, Maillard LT.
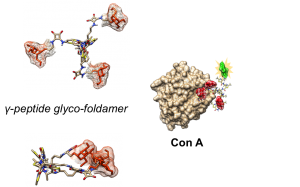
Abstract
Sugars play key roles in many molecular and cellular communication processes involving a family of proteins named lectins. The low affinity associated with sugar recognition is generally counterbalanced by the multivalent nature of the interaction. While many polyglycosylated architectures have been described, only a few studies focused on the impact of topology variations of the multivalent structures on the interaction with lectin proteins. One major interest of our group concerns the design of new highly predictable and stable molecular pseudo‐peptide architectures for therapeutic applications. In such a context, we described a class of constrained heterocyclic γ‐amino acids built around a thiazole ring, named ATCs. ATC oligomers are helical molecules resulting from the formation of a highly stable C9 hydrogen‐bonding pattern. Following our program, we herein address the potential of ATC oligomers as tunable scaffolds for the development of original multivalent glycoclusters.
