
Pascal VerdiÉ
Research Engineer, University of Montpellier
Pascal Verdié, a chemical engineer from the Ecole Nationale Supérieure de Chimie of Montpellier, France, performed a PhD program in Chemistry and Biology Sciences for Health, Montpellier University, which he graduated from in 2005. He worked on the development of selective ligands for the α-MSH MC1 receptor by the means of a huge program of peptide and combinatorial chemistry.
Pascal received a Research Associate position at Montpellier University since 2005. He’s in charge of the SynBio3 platform dedicated to assist the development of research programs in life science providing biomolecules and polymers of biological and pharmaceutical interest. He succeeded in obtaining the IBISA label since January 2013 and in achieving ISO 9001 certification in 2015.
Contact:
pascal.verdie@umontpellier.fr
ibmm-synbio3@umontpellier.fr
+33 (0)4 48 79 22 37
5 major publications :
«Structural Insights into Recognition of Chemokine Receptors by Staphylococcus Aureus Leukotoxins» P Lambey, O Otun, X Cong, F Hoh, L Brunel; P Verdié, C Grison, .; F Peysson, S Jeannot, T Durroux, C Bechara, S Granier, C Leyrat. eLife 2022, 11, e72555. https://doi.org/10.7554/eLife.72555.
«Active Targeted Nanoemulsions for Repurposing of Tegaserod in Alzheimer’s Disease Treatment» L Séguy, L Guyon, M Maurel, P Verdié, A Davis, S Corvaisier, V Lisowski, P Dallemagne, A-C Groo, A Malzert-Fréon. Pharmaceutics 2021, 13 (10), 1626. https://doi.org/10.3390/pharmaceutics13101626.
«Systemic Delivery of Tumor-Targeted Bax-Derived Membrane-Active Peptides for the Treatment of Melanoma Tumors in aHumanized SCID Mouse Model» A Karageorgis, M Claron, R Jugé, C Aspord, F Thoreau, C Leloup, J Kucharczak, J Plumas, M Henry, A Hurbin, P Verdié, J Martinez, G Subra, P Dumy, D Boturyn, A Aouacheria, and JL Coll1. Molecular Therapy, Molecular Therapy, 2017, 25, 2.
«Sol-gel synthesis of collagen-inspired peptide hydrogel», Echalier, S. Jebors, G. Laconde, L. Brunel, P. Verdie, L. Causse, A. Bethry, B. Legrand, H. Van Den Berghe, X. Garric, D. Noe, J. Martinez, A. Mehdi and G. Subra, Materials Today, 2017, 20, 2.
«Agonism, antagonism and inverse agonism bias at the Ghrelin receptor signaling» C M’Kadmi, J-P Leyris, L Onfroy, C Galés, A Saulière¶, D Gagne, M Damian, S Mary, M Maingot, S Denoyelle, P Verdié, J-A Fehrentz, J Martinez, J-L Banères, J Marie Journal of biological Chemistry, 2015, 290(45), 27021-27039.

Potent Lys Patch-Containing Stapled Peptides Targeting PCSK9
J Med Chem. 2021 Aug 12;64(15):10834-10848. doi: 10.1021/acs.jmedchem.0c02051. Epub 2021 Jul 15.
Bourbiaux K, Legrand B, Verdié P, Mallart S, Manette G, Minoletti C, Stepp JD, Prigent P, Le Bail JC
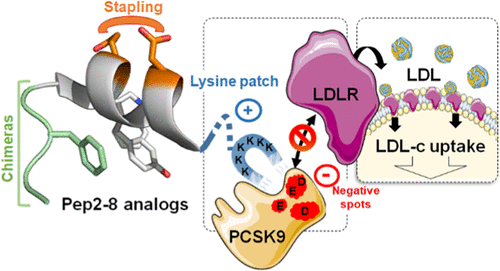
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9), identified as a regulator of low-density lipoprotein receptor (LDLR), plays a major role in cardiovascular diseases (CVD). Recently, Pep2-8, a small peptide with discrete three-dimensional structure, was found to inhibit the PCSK9/LDLR interaction. In this paper, we describe the modification of this peptide using stapled peptide and SIP technologies. Their combination yielded potent compounds such as 18 that potently inhibited the binding of PCSK9 to LDLR (KD = 6 ± 1 nM) and restored in vitro LDL uptake by HepG2 cells in the presence of PCSK9 (EC50 = 175 ± 40 nM). The three-dimensional structures of key peptides were extensively studied by circular dichroism and nuclear magnetic resonance, and molecular dynamics simulations allowed us to compare their binding mode to tentatively rationalize structure-activity relationships (SAR).

Site-specific grafting on titanium surfaces with hybrid temporin antibacterial peptides
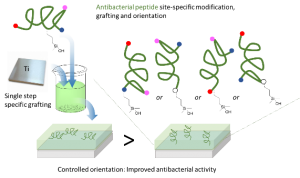
Abstract
Relying on a membrane-disturbing mechanism of action and not on any intracellular target, antimicrobial peptides (AMP) are attractive compounds to be grafted on the surface of implantable materials such as silicone catheters or titanium surgical implants. AMP sequences often display numerous reactive functions (e.g. amine, carboxylic acid) on their side chains and straightforward conjugation chemistries could lead to uncontrolled covalent grafting, random orientation, and non-homogenous density. To achieve an easy and site specific covalent attachment of unprotected peptides on titanium surfaces, we designed hybrid silylated biomolecules based on the temporin-SHa amphipathic helical antimicrobial sequence. With the grafting reaction being chemoselective, we designed five analogues displaying the silane anchoring function at the N-ter, C-ter or at different positions inside the sequence to get an accurate control of the orientation. Grafting density calculations were performed by XPS and the influence of the orientation of the peptide on the surface was clearly demonstrated by the measure of antimicrobial activity. Temporin amphipathic helices are described to permeabilize the bacterial membrane by interacting in a parallel orientation with it. Our results move in the direction of this mechanism as the selective grafting of hybrid temporin 2 through a lysine placed at the center of the peptide sequence, resulted in better biofilm growth inhibition of E. coli and S. epidermis than substrates in which temporins were grafted via their C- or N-terminus.
Pharmacological and Biochemical Characterization of TLQP-21 Activation of a Binding Site on CHO Cells
Front Pharmacol. 2017, Mar 30;8:167. doi: 10.3389/fphar.2017.00167. eCollection 2017.
Molteni L, Rizzi L, Bresciani E, Possenti R, Petrocchi Passeri P, Ghè C, Muccioli G, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, Biagini G, Binda A, Rivolta I, Locatelli V, Torsello A.
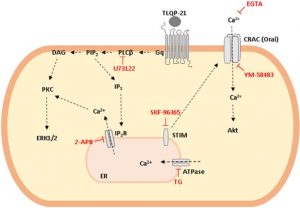
Abstract
VGF is a propeptide of 617 amino acids expressed throughout the central and the peripheral nervous system. VGF and peptides derived from its processing have been found in dense core vesicles and are released from neuronal and neuroendocrine cells via the regulated secretory pathway. Among VGF-derived neuropeptides, TLQP-21 (VGF556-576) has raised a huge interest and is one of most studied. TLQP-21 is a multifunctional neuropeptide involved in the control of several physiological functions, potentially including energy homeostasis, pain modulation, stress responsiveness and reproduction. Although little information is available about its receptor and the intracellular mechanisms mediating its biological effects, recent reports suggest that TLQP-21 may bind to the complement receptors C3aR1 and/or gC1qR. The first aim of this study was to ascertain the existence and nature of TLQP-21 binding sites in CHO cells. Secondly, we endeavored to characterize the ligand binding to these sites by using a small panel of VGF-derived peptides. And finally, we investigated the influence of TLQP-21 on selected intracellular signaling pathways. We report that CHO cells express a single class of saturable and specific binding sites for TLQP-21 with an affinity and capacity of Kd = 0.55 ± 0.05 × 10-9 M and Bmax = 81.7 ± 3.9 fmol/mg protein, respectively. Among the many bioactive products derived from the C-terminal region of VGF that we tested, TLQP-21 was the most potent in stimulating intracellular calcium mobilization in CHO cells; this effect is primarily due to its C-terminal fragment (HFHH-10). TLQP-21 induced rapid and transient dephosphorylation of phospholipase Cγ1 and phospholipase A2. Generation of IP3 and diacylglycerol was crucial for TLQP-21 bioactivity. In conclusion, our results suggest that the receptor stimulated by TLQP-21 belongs to the family of the Gq-coupled receptors, and its activation first increases membrane-lipid derived second messengers which thereby induce the mobilization of Ca2+ from the endoplasmic reticulum followed by a slower store-operated Ca2+ entry from outside the cell.
JMV5656, A Novel Derivative of TLQP-21, Triggers the Activation of a Calcium-Dependent Potassium Outward Current in Microglial Cells
Front Cell Neurosci. 2017 Feb 23;11:41. doi: 10.3389/fncel.2017.00041. eCollection 2017
Rivolta I, Binda A, Molteni L, Rizzi L, Bresciani E, Possenti R, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, Locatelli V, Torsello A.
Abstract
TLQP-21 (TLQPPASSRRRHFHHALPPAR) is a multifunctional peptide that is involved in the control of physiological functions, including feeding, reproduction, stress responsiveness, and general homeostasis. Despite the huge interest in TLQP-21 biological activity, very little is known about its intracellular mechanisms of action. In microglial cells, TLQP-21 stimulates increases of intracellular Ca2+ that may activate functions, including proliferation, migration, phagocytosis and production of inflammatory molecules. Our aim was to investigate whether JMV5656 (RRRHFHHALPPAR), a novel short analogue of TLQP-21, stimulates intracellular Ca2+ in the N9 microglia cells, and whether this Ca2+ elevation is coupled with the activation Ca2+-sensitive K+ channels. TLQP-21 and JMV5656 induced a sharp, dose-dependent increment in intracellular calcium. In 77% of cells, JMV5656 also caused an increase in the total outward currents, which was blunted by TEA (tetraethyl ammonium chloride), a non-selective blocker of voltage-dependent and Ca2+-activated potassium (K+) channels. Moreover, the effects of ion channel blockers charybdotoxin and iberiotoxin, suggested that multiple calcium-activated K+ channel types drove the outward current stimulated by JMV5656. Additionally, inhibition of JMV5656-stimulated outward currents by NS6180 (4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4 benzothiazin-3(4H)-one) and TRAM-34 (triarylmethane-34), indicated that KCa3.1 channels are involved in this JMV5656 mechanisms of action. In summary, we demonstrate that, in N9 microglia cells, the interaction of JMV5656 with the TLQP-21 receptors induced an increase in intracellular Ca2+, and, following extracellular Ca2+ entry, the opening of KCa3.1 channels.
Sol-gel synthesis of collagen-inspired peptide hydrogel
Materials Today , Pages: Ahead of Print, Journal, 2017, DOI: 10.1016/j.mattod.2017.02.001
C. Echalier, S. Jebors, G. Laconde, B. Guillaume, V. Luc, C. Pascal, B. Lea, A. Bethry, B. Legrand, H. Van Den Berghe, X. Garric, D. Noel, J. Martinez, A. Mehdi, G. Subra
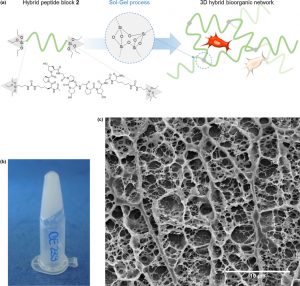
Abstract
Conceiving biomaterials able to mimic the specific environments of extracellular matrixes are a prerequisite for tissue engineering applications. Numerous types of polymers (PEG, PLA, etc.) have been used for the design of biocompatible scaffolds, but they are still less efficient than natural biopolymers such as collagen exts. Chem. modified and loaded with different bioactive factors, biopolymers afford an environment favorable to cell proliferation and differentiation. Unfortunately, they present several drawbacks, such as weak batch-to-batch reproducibility, potential immunogenicity and high cost of prodn. Herein we propose a fully synthetic covalent hydrogel obtained by sol-gel polymn. of a silylated peptide. We selected a short and low mol. building-block derived from the consensus collagen sequence [Pro-Hyp-Gly]. Interestingly, the sol-gel process occurs in physiol. buffer, enabling the embedment of stem cells. This collagen-inspired hydrogel provides a cell-friendly environment comparable to natural collagen substrates, demonstrating its potency as a biomimetic scaffold.
A switchable stapled peptide
Journal of Peptide Science, 2016, Volume: 22, Issue: 3, Pages: 143-148, DOI: 10.1002/psc.2851
A. Kalistratova, B. Legrand, P. Verdie, E. Naydenova, M. Amblard, J. Martinez, G. Subra
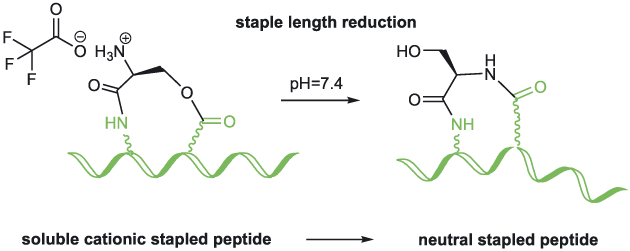
Abstract
The O-N acyl transfer reaction has gained significant popularity in peptide and medicinal chem. This reaction has been successfully applied to the synthesis of difficult sequence-contg. peptides, cyclic peptides, epimerization-free fragment coupling and more recently, to switchable peptide polymers. Herein, we describe a related strategy to facilitate the synthesis and purifn. of a hydrophobic stapled peptide. The staple consists of a serine linked through an amide bond formed from its carboxylic acid function and the side chain amino group of diaminopropionic acid and through an ester bond formed from its amino group and the side chain carboxylic acid function of aspartic acid. The α-amino group of serine was protonated during purifn. Interestingly, when the peptide was placed at physiol. pH, the free amino group initiated the O-N shift reducing the staple length by one atom, leading to a more hydrophobic stapled peptide.
Solid-supported synthesis and 5-HT7/5-HT1A receptor affinity of arylpiperazinylbutyl derivatives of 4,5-dihydro-1,2,4-triazine-6-(1H)-one
Chemical Biology & Drug Design, 2015, Volume: 86, Issue: 4, Pages: 697-703, DOI: 10.1111/cbdd.12539
K. Grychowska, N. Masurier, P. Verdie, G. Satala, A. J. Bojarski, J. Martinez, M. Pawlowski, G. Subra, P. Zajdel
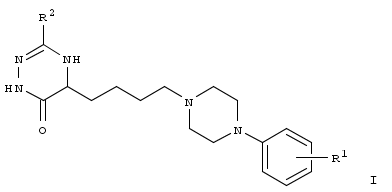
Abstract
A series of arylpiperazinylbutyl derivs. I [R1 = H, 2-OMe, 2-SMe, 2-F, 4-Cl, 2,3-diCl; R2 = Ph, 2-ClC6H4, cyclohexyl] of 4,5-dihydro-1,2,4-triazine-6(1H)-ones was designed and synthesized according to the new solid-supported methodol. In this approach, triazinone scaffold was constructed from the fmoc-protected glycine. The library representatives showed different levels of affinity for 5-HT7 and 5-HT1A receptors, among which compds. I [R1 = 2-SMe, 2-F; R2 = Ph and R1 = 2-OMe, 2-SMe, 2,3-diCl; R2 = cyclohexyl] were classified as dual 5-HT7/5-HT1A receptors ligands. The structure-affinity relationship anal. revealed that receptor affinity and selectivity of the tested compds. depended on the kind of substituent in position 3 of triazinone fragment as well as substitution pattern of phenylpiperazine moiety.
Agonism, Antagonism, and Inverse Agonism Bias at the Ghrelin Receptor Signaling
Journal of Biological Chemistry, 2015, Volume: 290, Issue: 45, Pages: 27021-27039, DOI: 10.1074/jbc.M115.659250
C. M’Kadmi, J.-P. Leyris, L. Onfroy, C. Gales, A. Sauliere, D. Gagne, M. Damian, S. Mary, M. Maingot, S. Denoyelle, P. Verdie, J.-A. Fehrentz, J. Martinez, J.-L. Baneres, J. Marie
Abstract
The G protein-coupled receptor GHS-R1a mediates ghrelin-induced growth hormone secretion, food intake, and reward-seeking behaviors. GHS-R1a signals through Gq, Gi/o, G13, and arrestin. Biasing GHS-R1a signaling with specific ligands may lead to the development of more selective drugs to treat obesity or addiction with minimal side effects. To delineate ligand selectivity at GHS-R1a signaling, we analyzed in detail the efficacy of a panel of synthetic ligands activating the different pathways assocd. with GHS-R1a in HEK293T cells. Besides β-arrestin2 recruitment and ERK1/2 phosphorylation, we monitored activation of a large panel of G protein subtypes using a bioluminescence resonance energy transfer-based assay with G protein-activation biosensors. We first found that unlike full agonists, Gq partial agonists were unable to trigger β-arrestin2 recruitment and ERK1/2 phosphorylation. Using G protein-activation biosensors, we then demonstrated that ghrelin promoted activation of Gq, Gi1, Gi2, Gi3, Goa, Gob, and G13 but not Gs and G12. Besides, we identified some GHS-R1a ligands that preferentially activated Gq and antagonized ghrelin-mediated Gi/Go activation. Finally, we unambiguously demonstrated that in addn. to Gq, GHS-R1a also promoted constitutive activation of G13. Importantly, we identified some ligands that were selective inverse agonists toward Gq but not of G13. This demonstrates that bias at GHS-R1a signaling can occur not only with regard to agonism but also to inverse agonism. Our data, combined with other in vivo studies, may facilitate the design of drugs selectively targeting individual signaling pathways to treat only the therapeutically relevant function.
Laser desorption ionization mass spectrometry of peptides on a hybrid CHCA organic-inorganic matrix
Journal of Proteomics, Volume, 2014, 108, Pages: 369-372, DOI: 10.1016/j.jprot.2014.06.005
C. Fleith, S. Cantel, G. Subra, A. Mehdi, J. Ciccione, J. Martinez, C. Enjalbal
Abstract
We report applications of new hybrid org.-inorg. silica based materials as laser desorption/ionization (LDI)-promoting surfaces for high-throughput identification of peptides. The driving force of our work was to design a new material composed of a conventional MALDI matrix covalently attached to silica with a high org./inorg. ratio in order to improve the UV absorption by such LDI hybrid matrixes. Amorphous CHCA-functionalized silica presenting an org. content up to 1.3 mmol g-1 (around 40% in wt. from TGA and elementary anal. measurements) gave very interesting LDI performances in terms of detection sensitivity as well as relative ionization discrepancy (spectral suppression) through the analyses of small synthetic peptide mixts. (550-1300 Da) taking CHCA and amorphous silica as model matrixes for control expts.
N- and O-acetylation of threonine residues in the context of proteomics
Journal of Proteomics, Volume, 2014, 108, Pages: 369-372, DOI: 10.1016/j.jprot.2014.06.005
J.-B. Boyer, A. Dedieu, J. Armengaud, P. Verdie, G. Subra, J. Martinez, C. Enjalbal
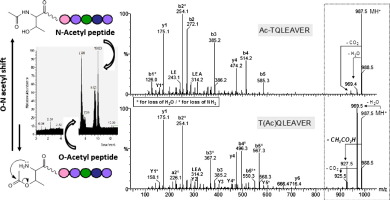
Abstract
The detection of post-translational modifications (PTMs) of proteins is a matter of intensive research. Among all possible pitfalls that may lead to misidentifications, the chem. stability of modified peptides is scarcely questioned. Global proteomic studies devoted to protein acetylation are becoming popular. Thus, we were concerned about the intrinsic stability of O-acetylated peptides because of the O-N acyl transfer reactivity occurring when an amino moiety is present in the vicinity of the acylated hydroxyl group. Here, the behavior of isomeric O- and N-acetylated, N-terminal threonine-contg. peptides was explored in a std. proteomic workflow. We demonstrated a strong chem. instability of O-acetylation, which prevents its detection.
