
JEAN Martinez
Professor, Faculty of Pharmacy, University OF MONTPELLIER
Jean Martinez received both his PhD in 1972 from the University of Montpellier 2, at the National School of Chemistry and his Dr Sciences Degree in 1976 from the same University, under the supervision of Professor F. Winternitz. The same year, he joined Dr E. Bricas group at Orsay, University of Paris Sud, as a post-doctorate fellow and in 1977 the laboratory of Professor M. Bodanszky at Case Western Reserve University in Cleveland, Ohio, USA, where he stayed till mid 1979.
Jean Martinez is a Full Professor at the University of Montpellier. In 2007, he created the « Institut des Biomolécules Max Mousseron » (IBMM), which he has been the Director until December 2014. He is actually Head of the department of Amino Acids, Peptides and Proteins at IBMM.
He served the University of Montpellier 1 as a Member of the Scientific Council for 8 years, and as Vice-President for 6 years (2009-2014).
Prof. Martinez is recognized for his important contributions, at the interface of chemistry and biology, to the development of methodology in organic and peptide synthesis, design and synthesis of various potent and selective neuropeptide analogues, and biomaterials containing biomolecules.
He has published over 900 original papers, 50 patents, numerous review articles, and he has been editor of several books. In 2012, he was accepted into the « Académie Nationale de Pharmacie », France, and in 2014 into the « Real Academia Nacional de Farmacia », Spain. In 2015, he has been nominated Docteur Honoris causa of the Jagellonian University of Kracow, Poland. He has received various prizes including the Leonidas Zervas Award (Barcelona, Spain 1990), the Silver Medal CNRS (Paris, France 1996), The Labbe Award (Paris, France 1996), The Mentzer Award (Caen, France 1998), the Max Bergmann Medal (Munster, Germany 2004), the Cathay Award (Shanghai, China 2006), the Akabori Award (Tokohama, Japan 2006), the Ehrlich Award (Lyon, France 2011), the Roche « Johannes Meinhofer Award » (Boulder Co, USA 2011), the Léon Velluz Award (Paris, France 2015), the Rudinger Award (Leipzig, Germany 2016). He is « Chevalier dans l’Ordre des Palmes Académiques » (2010), and Chevalier dans l’Ordre de la Légion d’Honneur (2011).
Jean Martinez served the European Peptide Society as French Representative, Member of the Executive Committee (1991-1998), Scientific Officer (1998-2001), and President (2001-2010).
Contact:
jean.martinez@umontpellier.fr
0033411759601
5 major publications :
1) “Turning peptide sequences into ribbon foldamers by a straightforward multicyclisation reaction“, V. Martin, B. Legrand, L. L. Vezenkov, M. Berthet, G. Subra, M. Calmes, J-L Bantignies, J. Martinez, & M. Amblard, Angew. Chem. Int. Ed., 54, 13966-13970 (2015). DOI: 10.1002/anie.201506955.
2) “A new way to silicon-based peptide polymers“, S. Jebors, J. Ciccione, S. Al Halifa, B. Nottelet, C. Enjalbal, C. M’Kadmi, M. Amblard, A. Mehdi, J. Martinez, & G. Subra, Angew. Chem. Int. Ed., 54, 3778-3782 (2015).
3) “Dipeptide mimic oligomer transporter mediates intracellular delivery of cathepsin D inhibitors: a potential target for cancer therapy”, M. Maynadier, L. Vezenkov, M. Amblard, V. Martin, C. Gandreuil, O. Vaillant, M. Gari-Bobo, I. Basile, J-F. Hernandez, M. Garcia, & J. Martinez, J. Controlled Release, 171, 251-257 (2013). DOI:10.1016/j.jconrel.2013.07.017.
4) “Ligands and signalling proteins govern the conformational landscape explored by a G-protein-coupled receptor“, S. Mary, M. Damian, M. Louet, N. Floquet, J-A Fehrentz, J. Marie, J. Martinez, & J-L Banères, Proc. Natl. Acad. Sci. USA, 109, 8304-8309 (2012).
5) “A straightforward approach for cellular-uptake quantification “, D. Paramelle, G. Subra, L. Vezenkov, M. Maynadier, C. André, C. Enjalbal, M. Calmes, M. Garcia, J. Martinez, & M. Amblard, Angew. Chem. Int. Ed.., 12, 747-753 (2010).

1,3-Diazepine: A Privileged Scaffold in Medicinal Chemistry
Med. Res. Rev., 2021, 41 (4), 2247-2315, https://doi.org/10.1002/med.21795
Y. Malki, J. Martinez, N. Masurier
Abstract
Privileged structures have been widely used as effective templates for drug discovery. While benzo-1,4-diazepine constitutes the first historical example of such a structure, the 1,3 analogue is just as rich in terms of applications in medicinal chemistry. The 1,3-diazepine moiety is present in numerous biological active compounds including natural products, and is used to design compounds displaying a large range of biological activities. It is present in the clinically used anticancer compound pentostatin, in several recent FDA approved β-lactamase inhibitors (e.g., avibactam) and also in coformycin, a natural product known as a ring-expanded purine analogue displaying antiviral and anticancer activities. Several other 1,3-diazepine containing compounds have entered into clinical trials. This heterocyclic structure has been and is still widely used in medicinal chemistry to design enzyme inhibitors, GPCR ligands, and so forth. This review endeavours to highlight the main use of the 1,3-diazepine scaffold and its derivatives, and their applications in medicinal chemistry, drug design, and therapy. We will focus more particularly on the development of enzyme inhibitors incorporating this scaffold, with a strong emphasis on the molecular interactions involved in the inhibition mechanism.
Neuropathic pain-alleviating activity of novel 5-HT6 receptor inverse agonists derived from 2-aryl-1H-pyrrole-3-carboxamide
Bioorg. Chem., 2021, 115, 105218, https://doi.org/10.1016/j.bioorg.2021.105218
Drop, F. Jacquot, V. Canale, S. Chaumont-Dubel, M. Walczak, G. Satała, K. Nosalska, G.U. Mahoro, K. Słoczyńska, K. Piska, S. Lamoine, E. Pękala, N. Masurier, A.J. Bojarski, M. Pawłowski, J. Martinez, G. Subra, X. Bantreil, F. Lamaty, A. Eschalier, P. Marin, C. Courteix, P. Zajdel
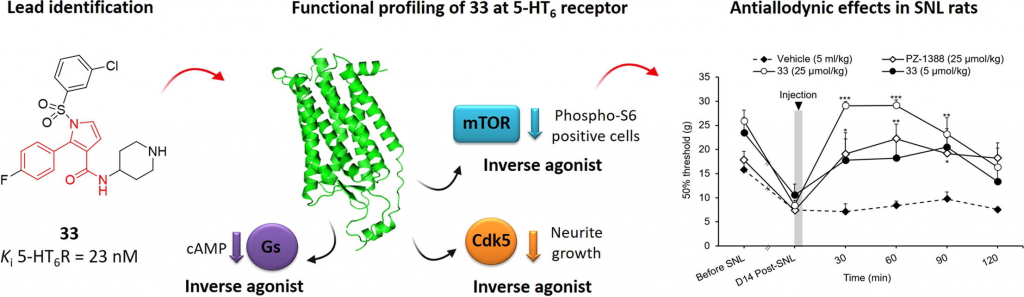
Abstract
The diverse signaling pathways engaged by serotonin type 6 receptor (5-HT6R) together with its high constitutive activity suggests different types of pharmacological interventions for the treatment of CNS disorders. Non-physiological activation of mTOR kinase by constitutively active 5-HT6R under neuropathic pain conditions focused our attention on the possible repurposing of 5-HT6R inverse agonists as a strategy to treat painful symptoms associated with neuropathies of different etiologies. Herein, we report the identification of compound 33 derived from the library of 2-aryl-1H-pyrrole-3-carboxamides as a potential analgesic agent. Compound 33 behaves as a potent 5-HT6R inverse agonist at Gs, Cdk5, and mTOR signaling. Preliminary ADME/Tox studies revealed preferential distribution of 33 to the CNS and placed it in the low-risk safety space. Finally, compound 33 dose-dependently reduced tactile allodynia in spinal nerve ligation (SNL)-induced neuropathic rats.

1-Aminobicyclo[2.2.2]octane-2-carboxylic Acid and Derivatives As Chiral Constrained Bridged Scaffolds for Foldamers and Chiral Catalysts
Acc Chem Res. 2021 Feb 2;54(3):685-696. doi: 10.1021/acs.accounts.0c00680. Epub 2021 Jan 19.
Milbeo P, Martinez J, Amblard M, Calmès M, Legrand B
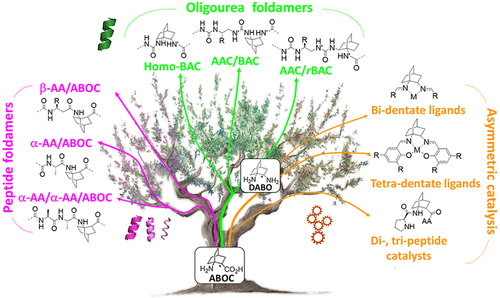
Abstract
The improvement of molecular diversity is one of the major concerns of chemists since the continuous development of original synthetic molecules provides unique scaffolds usable in organic and bioorganic chemistry. The challenge is to develop versatile platforms with highly controlled chemical three-dimensional space thanks to controlled chirality and conformational restraints. In this respect, cyclic β-amino acids are of great interest with applications in various fields of chemistry. In addition to their intrinsic biological properties, they are important precursors for the synthesis of new generations of bioactive compounds such as antibiotics, enzyme inhibitors, and antitumor agents. They have also been involved in asymmetric synthesis as efficient organo-catalysts in their free form and as derivatives. Finally, constrained cyclic β-amino acids have been incorporated into oligomers to successfully stabilize original structures in foldamer science with recent successes in health, material science, and catalysis. Over the last ∼10 years, we focused on bicyclic β-amino acids possessing a bicyclo[2.2.2]octane structure. This latter is a structural key element in numerous families of biologically active natural and synthetic products and is an interesting template for asymmetric synthesis. Nonetheless, reported studies on bicyclic carbo-bridged compounds are rather limited compared to those on bicyclic-fused and heterobridged derivatives. In this Account, we particularly focused on the synthesis and applications of the 1-aminobicyclo[2.2.2]octane-2-carboxylic acid, named, ABOC, and its derivatives. This highly constrained bicyclic β-amino acid, with a sterically hindered bridgehead primary amine and an endocyclic chiral center, displays drastically reduced conformational freedom. In addition, its high bulkiness strongly impacts the spatial orientation of the appended functionalities and the conformation of adjacent building blocks. Thus, we have first expanded a fundamental synthetic work by a wide ranging study in the field of foldamers, in the design of various stable peptide/peptidomimetic helical structures incorporating the ABOC residue (11/9-, 18/16-, 12/14/14-, and 12/10-helices). In addition, such bicyclic residue was fully compatible with and stabilized the canonical oligourea helix, whereas very few cyclic β-amino acids have been incorporated into oligoureas. In addition, we have pursued with the synthesis of some ABOC derivatives, in particular the 1,2-diaminobicyclo[2.2.2]octane chiral diamine, named DABO, and its investigation in chiral catalytic systems. Covalent organo-catalysis of the aldol reaction using ABOC-containing tripeptide catalysts provided a range of aldol products with high enantioselectivity. Moreover, the double reductive condensation of DABO with various aldehydes allowed the building of new chiral ligands that proved their efficiency in the copper-catalyzed asymmetric Henry reaction.
Turning peptides into bioactive nylons
European Polymer Journal 2020. doi: 10.1016/j.eurpolymj.2020.109886
Said Jebors, Coline Pinese, Titouan Montheil, Audrey Bethry, Simon Verquin, Louise Plais, Marie Moulin, Chloé Dupont, Xavier Garric, Ahmad Mehdi, Jean Martineza and Gilles Subra.
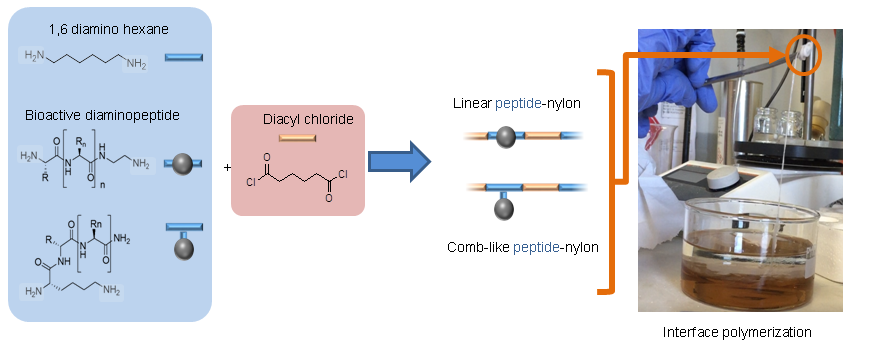
Abstract
New synthetic textiles with physical and/or biological properties are increasingly used in medical applications.While a simple textile coating is usually carried out to obtain biological properties, covalent grafting should be considered for long-term applications. Herein, we have developed a new hybrid bioactive nylon whose synthesis involves a peptide sequence with a diacyl derivative. Numerous types of peptide-nylons were prepared by varying the molar percentage (0.1 %, 1 % and 10%) and orientation of the peptide in the polymer backbone. Nylons incorporating antibacterial peptides significantly inhibited S. aureus proliferation whereas nylons functionalized with cell-adhesive peptide enhanced the proliferation of L929 fibroblast. These results show that the incorporation of the peptides directly into the nylon skeleton is efficient and provides biological properties that suggest new ways of functionalizing biomedical textiles
Inorganic Sol–Gel Polymerization for Hydrogel Bioprinting.
ACS Omega 2020, 5, 6, 2640-2647. doi: 10.1021/acsomega.9b03100.
Montheil T, Maumus M, Valot L, Lebrun A, Martinez J, Amblard M, Noël D, Mehdi A, Subra G.
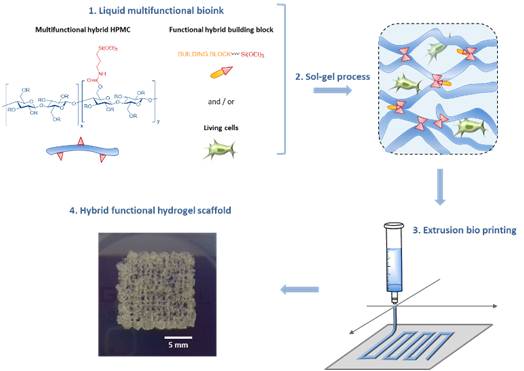
Abstract
An inorganic sol–gel polymerization process was used as a cross-linking reaction during three-dimensional (3D) bioprinting of cell-containing hydrogel scaffolds. Hybrid hydroxypropyl methyl cellulose (HPMC), with a controlled ratio of silylation, was prepared and isolated as a 3D-network precursor. When dissolved in a biological buffer containing human mesenchymal stem cells, it yields a bioink that can be printed during polymerization by extrusion. It is worth noting that the sol–gel process proceeded at pH 7.4 using biocompatible mode of catalysis (NaF and glycine). The printing window was determined by rheology and viscosity measurements. The physicochemical properties of hydrogels were studied. Covalent functionalization of the network can be easily performed by adding a triethoxysilyl-containing molecule; a fluorescent hybrid molecule was used as a proof of concept.
Biocompatible Glycine-Assisted Catalysis of the Sol-Gel Process: Development of Cell-Embedded Hydrogels.
Chempluschem. 2019 Nov;84(11):1720-1729. doi: 10.1002/cplu.201900509.
Valot L, Maumus M, Montheil T, Martinez J, Noël D, Mehdi A, Subra G.
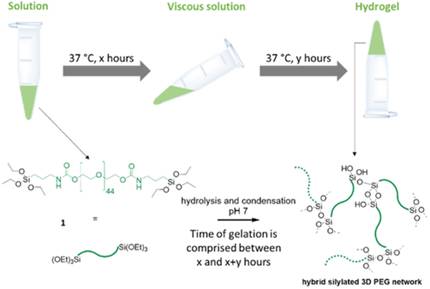
Abstract
The sol‐gel process can be used for hydrogel cross‐linking, thus opening an attractive route for the design of biocompatible hydrogels under soft conditions. The sol‐gel process can be catalysed at basic or acidic pH values, under neutral conditions with the addition of a nucleophile. Therefore, working around pH 7 unlocks the possibility of direct cell embedment and the preparation of bioinks. We aimed to propose a generic method for sol‐gel 3D bioprinting, and first screened different nucleophilic catalysts using bis‐silylated polyethylene glycol (PEG) as a model hydrogel. A synergistic effect of glycine and NaF, used in low concentrations to avoid any toxicity, was observed. Biocompatibility of the approach was demonstrated by embedding primary mouse mesenchymal stem cells. The measure of viscosity as a function of time showed the impact of reaction parameters, such as temperature, complexity of the medium, pH and cell addition, on the kinetics of the sol‐gel process, and allowed prediction of the gelation time.
Chemical cross-linking methods for cell encapsulation in hydrogels
Mater Today Comm., 2019, 20, 100536. doi: 10.1016/j.mtcomm.2019.05.012
Echalier C, Valot L, Martinez J, Mehdi A and Subra G.

Abstract
Cell-encapsulating hydrogels are of tremendous interest in regenerative medicine. Tissue engineering relies on biomaterials able to act as artificial extracellular matrices to guide cells towards the development of new tissues. Therefore, considerable efforts have been made to design biomaterials which mimic cells’ native environment, thus encouraging natural behavior. The choice of biomaterial in which cells are embedded is crucial for their survival, proliferation and differentiation. Being more stable, chemical hydrogels are preferred over physical hydrogels as cell-laden substrates. When designing chemical hydrogels, scientists must choose not only the nature of the network (synthetic and/or bio-polymers) but also the type of cross-link bridging hydrogel constituents. For that purpose, numerous chemistries have been used (i) to introduce reactive functions on the hydrogel precursors and (ii) to form covalent bonds in the presence of living cells. The review will discuss the advantages and limitations of each strategy.
Chemical insights into bioinks for 3D printing
Chem. Soc. Rev., 2019, 48, 15, 4049-4086 doi: 10.1039/C7CS00718C
Valot L, Martinez J, Mehdi A and Subra G.
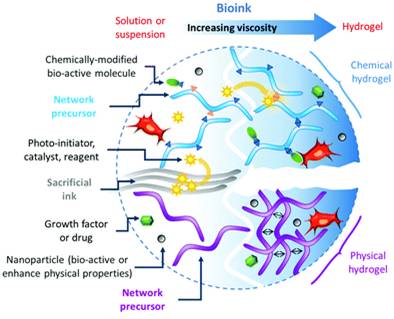
Abstract
3D printing has triggered the acceleration of numerous research areas in health sciences, which traditionally used cells as starting materials, in particular tissue engineering, regenerative medicine and also in the design of more relevant bioassays for drug discovery and development. While cells can be successfully printed in 2D layers without the help of any supporting biomaterial, the obtainment of more complex 3D architectures requires a specific bioink, i.e. a material in which the cells are embedded during and after the printing process helping to support them while they are arranged in superimposed layers. The bioink plays a critical role in bioprinting: first, it must be adapted to the 3D printing technology; then, it must fulfil the physicochemical and mechanical characteristics of the target construct (e.g. stiffness, elasticity, robustness, transparency); finally it should guarantee cell viability and eventually induce a desired behaviour. This review focuses on the nature of bioink components of natural or synthetic origin, and highlights the chemistry required for the establishment of the 3D network in conditions compatible with the selected 3D printing technique and cell survival.
Self-mineralization and assembly of a bis-silylated Phe–Phe pseudodipeptide to a structured bioorganic–inorganic material
Mater. Horiz., 2019, 6, 2040-2046 doi: 10.1039/C9MH00580C
Jebors S, Valot L, Echalier C, Legrand L, Mikhaleff R,Van Der Lee A, Arenal R, Dumy P, Amblard M, Martinez J, Mehdi A and Subra G.
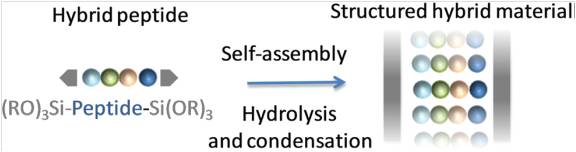
Abstract
Self-mineralization of a trialkoxysilyl hybrid peptide yields in a single step a nanostructured hybrid material. A bis-silylated pseudodipeptide inspired from the Phe–Phe dipeptide was used to program the assembly by sol–gel polymerization under heterogeneous conditions, in water at pH 1.5 without any structure-directing agent. A mechanism deciphering the hybrid material assembly was proposed thanks to 1H NMR spectroscopy. First, water-insoluble hybrid building blocks were hydrolysed into their soluble silanol counterparts. Then, these transitional species, thanks to hydrogen bonding and π–π stacking, self-assembled in solution. Last, the proximity of the silanol moieties favoured their polycondensation into growing siloxane oligomers, which spontaneously precipitated to produce an ordered hybrid material.
How are 1,2,3-triazoles accommodated in helical secondary structures?
Org Biomol Chem. 2018 May 15. doi: 10.1039/c8ob00686e
Ben Haj Salah K, Das S , Ruiz N , Andreu V , Martinez J , Wenger E , Amblard M , Didierjean C , Legrand B , Inguimbert N
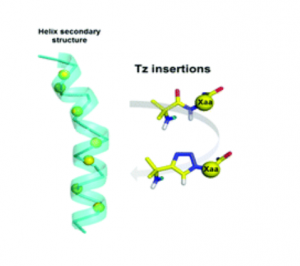
Abstract
1,4-Disubstituted-1,2,3-triazole (Tz) is widely used in peptides as a trans-amide bond mimic, despite having hazardous effects on the native peptide activity. The impact of amide bond substitution by Tz on peptide secondary structures is scarcely documented. We performed a Tz scan, by systematically replacing peptide bonds following the Aib residues with Tz on two model peptaibols: alamethicin F50/5 and bergofungin D, which adopt stable α- and 310 helices, respectively. We observed that the Tz insertion, whatever its position in the peptide sequences, abolished their antimicrobial activity. The structural consequences of this insertion were further investigated using CD, NMR and X-ray diffraction. Importantly, five crystal structures that were incorporated with Tz were solved, showing various degrees of alteration of the helical structures, from minor structural perturbation of the helix to partial disorder. Together, these results showed that Tz insertions impair helical secondary structures.
12/10-Helix in Mixed β-Peptides Alternating Bicyclic and Acyclic β-Amino Acids: Probing the Relationship between Bicyclic Side Chain and Helix Stability
Chemistry. 2018 Dec 12. doi: 10.1002/chem.201804404. Epub 2018 Nov 15
Simon M, Milbeo P, Liu H, André C, Wenger E, Martinez J, Amblard M, Aubert E, Legrand B, Calmès M.
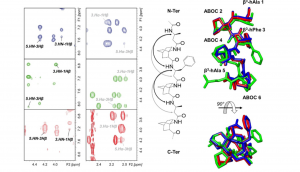
Abstract
12/10-Helices constitute suitable templates that can be used to design original structures. Nevertheless, they often suffer from a weak stability in polar solvents because they exhibit a mixed hydrogen-bond network resulting in a small macrodipole. In this work, stable and functionalizable 12/10-helices were developed by alternating a highly constrained β2, 3, 3 -trisubstituted bicyclic amino acid (S)-1-aminobicyclo[2.2.2]octane-2-carboxylic acid ((S)-ABOC) and an acyclic substituted β-homologated proteinogenic amino acid (l-β3 -hAA). Based on NMR spectroscopic analysis, it was shown that such mixed β-peptides display well-defined right-handed 12/10-helices in polar, apolar, and chaotropic solvents; that are, CD3 OH, CDCl3 , and [D6 ]DMSO, respectively. The stability of the hydrogen bonds forming the C10 and C12 pseudocycles as well as the benefit provided by the use of the constrained bicyclic ABOC versus typical acyclic β-amino acids sequences when designing 12/10-helix were investigated using NH/ND NMR exchange experiments and DFT calculations in various solvents. These studies showed that the β3 -hAA/(S)-ABOC helix displayed a more stable hydrogen-bond network through specific stabilization of the C10 pseudocycles involving the bridgehead NH of the ABOC bicyclic scaffold.
Site-specific grafting on titanium surfaces with hybrid temporin antibacterial peptides
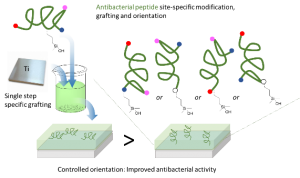
Abstract
Relying on a membrane-disturbing mechanism of action and not on any intracellular target, antimicrobial peptides (AMP) are attractive compounds to be grafted on the surface of implantable materials such as silicone catheters or titanium surgical implants. AMP sequences often display numerous reactive functions (e.g. amine, carboxylic acid) on their side chains and straightforward conjugation chemistries could lead to uncontrolled covalent grafting, random orientation, and non-homogenous density. To achieve an easy and site specific covalent attachment of unprotected peptides on titanium surfaces, we designed hybrid silylated biomolecules based on the temporin-SHa amphipathic helical antimicrobial sequence. With the grafting reaction being chemoselective, we designed five analogues displaying the silane anchoring function at the N-ter, C-ter or at different positions inside the sequence to get an accurate control of the orientation. Grafting density calculations were performed by XPS and the influence of the orientation of the peptide on the surface was clearly demonstrated by the measure of antimicrobial activity. Temporin amphipathic helices are described to permeabilize the bacterial membrane by interacting in a parallel orientation with it. Our results move in the direction of this mechanism as the selective grafting of hybrid temporin 2 through a lysine placed at the center of the peptide sequence, resulted in better biofilm growth inhibition of E. coli and S. epidermis than substrates in which temporins were grafted via their C- or N-terminus.
GHSR-D2R heteromerization modulates dopamine signaling through an effect on G protein conformation
Proc Natl Acad Sci U S A. 2018 Apr 24;115(17):4501-4506. doi: 10.1073/pnas.1712725115. Epub 2018 Apr 9
Damian M, Pons V, Renault P, M’Kadmi C, Delort B, Hartmann L, Kaya AI, Louet M, Gagne D, Ben Haj Salah K, Denoyelle S, Ferry G, Boutin JA, Wagner R, Fehrentz JA, Martinez J, Marie J, Floquet N, Galès C, Mary S, Hamm HE, Banères JL.
Abstract
IThe growth hormone secretagogue receptor (GHSR) and dopamine receptor (D2R) have been shown to oligomerize in hypothalamic neurons with a significant effect on dopamine signaling, but the molecular processes underlying this effect are still obscure. We used here the purified GHSR and D2R to establish that these two receptors assemble in a lipid environment as a tetrameric complex composed of two each of the receptors. This complex further recruits G proteins to give rise to an assembly with only two G protein trimers bound to a receptor tetramer. We further demonstrate that receptor heteromerization directly impacts on dopamine-mediated Gi protein activation by modulating the conformation of its α-subunit. Indeed, association to the purified GHSR:D2R heteromer triggers a different active conformation of Gαi that is linked to a higher rate of GTP binding and a faster dissociation from the heteromeric receptor. This is an additional mechanism to expand the repertoire of GPCR signaling modulation that could have implications for the control of dopamine signaling in normal and physiopathological conditions.
Inorganic polymerization: an attractive route to biocompatible hybrid hydrogels
J. Mater. Chem. B, 2018,6, 3434-3448, doi 10.1039/C8TB00456K
T
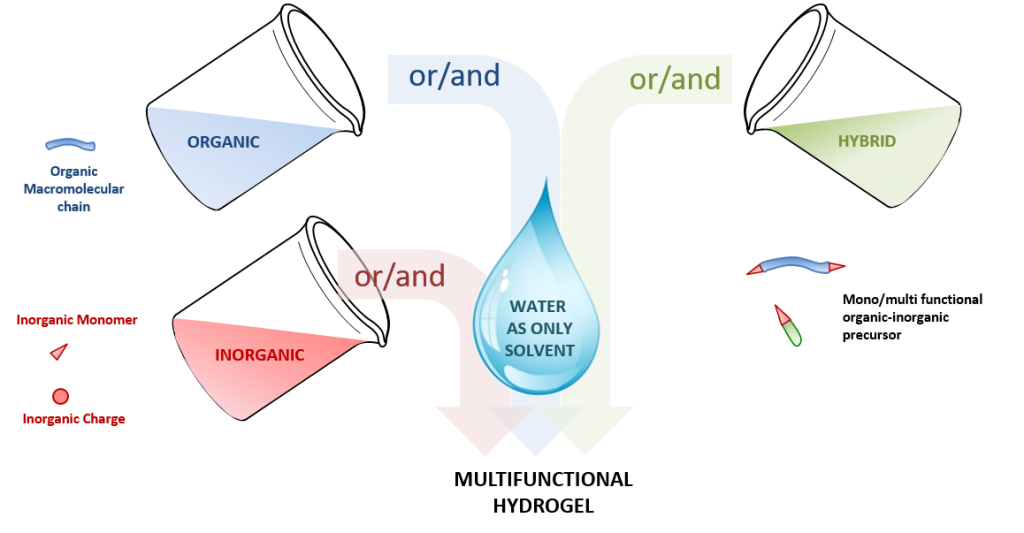
Abstract
As an intermediate state between liquid and solid materials, hydrogels display unique properties, opening a wide scope of applications, especially in the biomedical field. Organic hydrogels are composed of an organic network cross-linked via chemical or physical reticulation nodes. In contrast, hybrid hydrogels are defined by the coexistence of organic and inorganic moieties in water. Inorganic polymerization, i.e. the sol–gel process, is one of the main techniques leading to hybrid hydrogels. The chemoselectivity of this method proceeds through hydrolysis and condensation reactions of metal oxide moieties. In addition, the mild reaction conditions make this process very promising for the preparation of water-containing materials and their bio-applications.
Heteromultivalent targeting of integrin αvβ3 and neuropilin 1 promotes cell survival via the activation of the IGF-1/insulin receptors
Biomaterials. 2018 Feb;155:64-79. doi: 10.1016/j.biomaterials.2017.10.042. Epub 2017 Oct 29.
Jia T, Choi J, Ciccione J, Henry M, Mehdi A, Martinez J, Eymin B, Subra G, Coll JL.
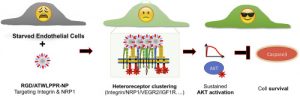
Abstract
Angiogenesis strongly depends on the activation of integrins, especially integrin αvβ3, and of neuropilin-1 (NRP-1), a co-receptor of VEGFR2. Dual-targeted molecules that simultaneously block both of them are expected have increased anti-angiogenic and antitumor activity. Toward this goal, we generated bifunctional 40 nm-sized silica nanoparticles (NPs) coated with controlled amounts of cRGD and ATWLPPR peptides and studied their affinity, selectivity and biological activity in HUVECs. Sub-nanomolar concentrations of NPs grafted either with ATWLPPR alone or in combination with cRGD exhibit potent and specific antagonist activity against VEGFR2/AKT signaling. However, a 1 nM concentration of the cRGD/ATWLPPR-heteromultivalent particles (RGD/ATW-NPs) also blocks the phosphorylation of VEGFR2 while co-inducing an unexpected long-lasting activation of AKT via IGF-1R/IR-AKT/GSK3β/eNOS signaling that stimulates cell survival and abrogates the intrinsic toxicity of silica-NPs to serum-starved HUVECs. We also showed that their repeated intravenous administration was associated with the proliferation of human U87MG tumor cells engrafted in nude mice and a dilatation of the tumor blood vessels. We present biochemical evidence for the complex cross-talk generated by the binding of the heteromultivalent NPs with αvβ3-integrin and with NRP1. In particular, we show for the first time that such heteromultivalent NPs can trans-activate IGF-1/insulin receptors and exert dose-dependent pro-survival activity. This study demonstrates the difficulties in designing targeted silica-based NPs for antiangiogenic therapies and the possible risks posed by undesirable side effects.
The GHR-R antagonist JMV 2959 neither induces malaise nor alters the malaise property of LiCl in the adult male rat
Physiol Behav. 2018 Jan 1;183:46-48. doi: 10.1016/j.physbeh.2017.10.017. Epub 2017 Oct 19.
Rodriguez JA, Fehrentz JA, Martinez J, Ben Haj Salah K, Wellman PJ.
Abstract
The orexigenic peptide ghrelin (GHR) interacts with ghrelin receptors (GHR-Rs) to modulate brain reinforcement and feeding circuits. Pharmacological inactivation of GHR-Rs via administration of the drug JMV 2959 attenuates the rewarding/reinforcing effects of several drugs of abuse including alcohol, morphine, amphetamine and nicotine. One view of these results is that inactivation of GHR-Rs taps into brain reinforcement/feeding circuits acted upon by drugs of abuse. An alternate explanation is that JMV 2959 may induce malaise, which in turn may limit reinforcement as well as food ingestion. This is a variable of interest given that nicotine alone can induce malaise which may be enhanced by JMV 2959. In the present study, we assessed the capacity of JMV 2959 to produce malaise using a conditioned taste aversion (CTA) task. Adult male rats were allowed to consume a 0.1% sodium saccharin solution and then injected IP with either vehicle, 0.4mg/kg nicotine, 3mg/kg JMV 2959, a combination of 0.4mg/kg nicotine and 3mg/kg JMV 2959, or 32mg/kg lithium chloride (a positive control known to support induction of CTA). Lithium chloride produced a robust avoidance of the saccharin solution in subsequent 2 bottle (water and saccharin) tests, whereas JMV 2959 alone did not induce CTA. The combination of JMV 2959 and nicotine induced a moderate degree of CTA that was similar to that produced by nicotine alone. These results suggest that JMV 2959 is unlikely to limit either reinforcement or food ingestion via induction of malaise.
Growth hormone secretagogues hexarelin and JMV2894 protect skeletal muscle from mitochondrial damages in a rat model of cisplatin-induced cachexia
Sci Rep. 2017 Oct 12;7(1):13017. doi: 10.1038/s41598-017-13504-y.
Sirago G, Conte E, Fracasso F, Cormio A, Fehrentz JA, Martinez J, Musicco C, Camerino GM, Fonzino A, Rizzi L, Torsello A, Lezza AMS, Liantonio A, Cantatore P, Pesce V.
Abstract
Chemotherapy can cause cachexia, which consists of weight loss associated with muscle atrophy. The exact mechanisms underlying this skeletal muscle toxicity are largely unknown and co-therapies to attenuate chemotherapy-induced side effects are lacking. By using a rat model of cisplatin-induced cachexia, we here characterized the mitochondrial homeostasis in tibialis anterior cachectic muscle and evaluated the potential beneficial effects of the growth hormone secretagogues (GHS) hexarelin and JMV2894 in this setting. We found that cisplatin treatment caused a decrease in mitochondrial biogenesis (PGC-1α, NRF-1, TFAM, mtDNA, ND1), mitochondrial mass (Porin and Citrate synthase activity) and fusion index (MFN2, Drp1), together with changes in the expression of autophagy-related genes (AKT/FoxO pathway, Atg1, Beclin1, LC3AII, p62) and enhanced ROS production (PRX III, MnSOD). Importantly, JMV2894 and hexarelin are capable to antagonize this chemotherapy-induced mitochondrial dysfunction. Thus, our findings reveal a key-role played by mitochondria in the mechanism responsible for GHS beneficial effects in skeletal muscle, strongly indicating that targeting mitochondrial dysfunction might be a promising area of research in developing therapeutic strategies to prevent or limit muscle wasting in cachexia.
Involvement of PPARγ in the Anticonvulsant Activity of EP-80317, a Ghrelin Receptor Antagonist
Front Pharmacol. 2017 Sep 22;8:676. doi: 10.3389/fphar.2017.00676. eCollection 2017.
Lucchi C, Costa AM, Giordano C, Curia G, Piat M, Leo G, Vinet J, Brunel L, Fehrentz JA, Martinez J, Torsello A, Biagini G.
Abstract
Ghrelin, des-acyl ghrelin and other related peptides possess anticonvulsant activities. Although ghrelin and cognate peptides were shown to physiologically regulate only the ghrelin receptor, some of them were pharmacologically proved to activate the peroxisome proliferator-activated receptor gamma (PPARγ) through stimulation of the scavenger receptor CD36 in macrophages. In our study, we challenged the hypothesis that PPARγ could be involved in the anticonvulsant effects of EP-80317, a ghrelin receptor antagonist. For this purpose, we used the PPARγ antagonist GW9662 to evaluate the modulation of EP-80317 anticonvulsant properties in two different models. Firstly, the anticonvulsant effects of EP-80317 were studied in rats treated with pilocarpine to induce status epilepticus (SE). Secondly, the anticonvulsant activity of EP-80317 was ascertained in the repeated 6-Hz corneal stimulation model in mice. Behavioral and video electrocorticographic (ECoG) analyses were performed in both models. We also characterized levels of immunoreactivity for PPARγ in the hippocampus of 6-Hz corneally stimulated mice. EP-80317 predictably antagonized seizures in both models. Pretreatment with GW9662 counteracted almost all EP-80317 effects both in mice and rats. Only the effects of EP-80317 on power spectra of ECoGs recorded during repeated 6-Hz corneal stimulation were practically unaffected by GW9662 administration. Moreover, GW9662 alone produced a decrease in the latency of tonic-clonic seizures and accelerated the onset of SE in rats. Finally, in the hippocampus of mice treated with EP-80317 we found increased levels of PPARγ immunoreactivity. Overall, these results support the hypothesis that PPARγ is able to modulate seizures and mediates the anticonvulsant effects of EP-80317.
C9/12 Ribbon-Like Structures in Hybrid Peptides Alternating α- and Thiazole-Based γ-Amino Acids
Chemistry. 2017 Dec 11;23(69):17584-17591. doi: 10.1002/chem.201704001. Epub 2017 Nov 15.
Bonnel C, Legrand B, Simon M, Martinez J, Bantignies JL, Kang YK, Wenger E, Hoh F, Masurier N, Maillard LT.
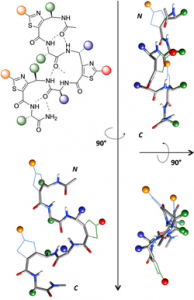
Abstract
According to their restricted conformational freedom, heterocyclic γ-amino acids are usually considered to be related to Z-vinylogous γ-amino acids. In this context, oligomers alternating α-amino acids and thiazole-based γ-amino acids (ATCs) were expected to fold into a canonical 12-helical shape as described for α/γ-hybrid peptides composed of cis-α/β-unsaturated γ-amino acids. However, through a combination of X-ray crystallography, NMR spectroscopy, FTIR experiments, and DFT calculations, it was determined that the folding behavior of ATC-containing hybrid peptides is much more complex. The homochiral α/(S)-ATC sequences were unable to adopt a stable conformation, whereas the heterochiral α/(R)-ATC peptides displayed novel ribbon structures stabilized by unusual C9/12 -bifurcated hydrogen bonds. These ribbon structures could be considered as a succession of pre-organized γ/α dipeptides and may provide the basis for designing original α-helix mimics.
Ribbon-like Foldamers for Cellular Uptake and Drug Delivery
Chembiochem 2017 Nov 2;18(21):2110-2114. doi: 10.1002/cbic.201700455. Epub 2017 Sep 22.
Vezenkov LL, Martin V, Bettache N, Simon M, Messerschmitt A, Legrand B, Bantignies JL, Subra G, Maynadier M, Bellet V, Garcia M, Martinez J, Amblard M.
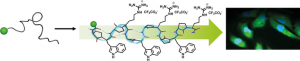
Abstract
Different intracellular delivery systems of bioactive compounds have been developed, including cell-penetrating peptides. Although usually nontoxic and biocompatible, these vectors share some of the general drawbacks of peptides, notably low bioavailability and susceptibility to protease degradation, that limit their use. Herein, the conversion of short peptide sequences into poly-α-amino-γ-lactam foldamers that adopt a ribbon-like structure is investigated. This template is used to distribute critical cationic and/or hydrophobic groups on both sides of the backbone, leading to potent short, cell-permeable foldamers with a low positive-charge content. The lead compound showed dramatically improved protease resistance and was able to efficiently deliver a biologically relevant cargo inside cells. This study provided a simple strategy to convert short peptide sequences into efficient protease-resistant cell-penetrating foldamers.
Microgels of silylated HPMC as a multimodal system for drug co-encapsulation
Int J Pharm. 2017 Nov 5;532(2):790-801. doi: 10.1016/j.ijpharm.2017.07.074. Epub 2017 Jul 27.
Zayed M, Tourne-Peteilh C, Ramonda M, Rethore G, Weiss P, Martinez J, Subra G, Mehdi A, Devoisselle JM, Legrand P.
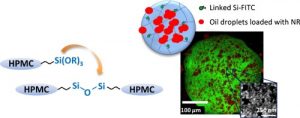
Abstract
Combined therapy is a global strategy developed to prevent drug resistance in cancer and infectious diseases. In this field, there is a need of multifunctional drug delivery systems able to co-encapsulate small drug molecules, peptides, proteins, associated to targeting functions, nanoparticles. Silylated hydrogels are alkoxysilane hybrid polymers that can be engaged in a sol-gel process, providing chemical cross linking in physiological conditions, and functionalized biocompatible hybrid materials. In the present work, microgels were prepared with silylated (hydroxypropyl)methyl cellulose (Si-HPMC) that was chemically cross linked in soft conditions of pH and temperature. They were prepared by an emulsion templating process, water in oil (W/O), as microreactors where the condensation reaction took place. The ability to functionalize the microgels, so-called FMGs, in a one-pot process, was evaluated by grafting a silylated hydrophilic model drug, fluorescein (Si-Fluor), using the same reaction of condensation. Biphasic microgels (BPMGs) were prepared to evaluate their potential to encapsulate lipophilic model drug (Nile red). They were composed of two separate compartments, one oily phase (sesame oil) trapped in the cross linked Si-HPMC hydrophilic phase. The FMGs and BPMGs were characterized by different microscopic techniques (optic, epi-fluorescence, Confocal Laser Scanning Microscopy and scanning electronic microscopy), the mechanical properties were monitored using nano indentation by Atomic Force Microscopy (AFM), and different preliminary tests were performed to evaluate their chemical and physical stability. Finally, it was demonstrated that it is possible to co-encapsulate both hydrophilic and hydrophobic drugs, in silylated microgels, that were physically and chemically stable. They were obtained by chemical cross linking in soft conditions, and without surfactant addition during the emulsification process. The amount of drug loaded was in favor of further biological activity. Mechanical stimulations should be necessary to trigger drug release.
1,2,4-Triazole-3-thione Compounds as Inhibitors of Dizinc Metallo-β-lactamases
ChemMedChem. 2017 Jun 21;12(12):972-985. doi: 10.1002/cmdc.201700186. Epub 2017 Jun 12.
Sevaille L, Gavara L, Bebrone C, De Luca F, Nauton L, Achard M, Mercuri P, Tanfoni S, Borgianni L, Guyon C, Lonjon P, Turan-Zitouni G, Dzieciolowski J, Becker K, Bénard L, Condon C, Maillard L, Martinez J, Frère JM, Dideberg O, Galleni M, Docquier JD, Hernandez JF.
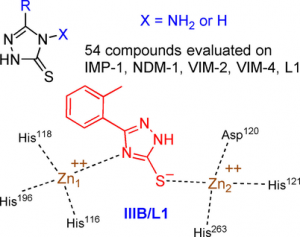
Abstract
Metallo-β-lactamases (MBLs) cause resistance of Gram-negative bacteria to β-lactam antibiotics and are of serious concern, because they can inactivate the last-resort carbapenems and because MBL inhibitors of clinical value are still lacking. We previously identified the original binding mode of 4-amino-2,4-dihydro-5-(2-methylphenyl)-3H-1,2,4-triazole-3-thione (compound IIIA) within the dizinc active site of the L1 MBL. Herein we present the crystallographic structure of a complex of L1 with the corresponding non-amino compound IIIB (1,2-dihydro-5-(2-methylphenyl)-3H-1,2,4-triazole-3-thione). Unexpectedly, the binding mode of IIIB was similar but reverse to that of IIIA. The 3 D structures suggested that the triazole-thione scaffold was suitable to bind to the catalytic site of dizinc metalloenzymes. On the basis of these results, we synthesized 54 analogues of IIIA or IIIB. Nineteen showed IC50 values in the micromolar range toward at least one of five representative MBLs (i.e., L1, VIM-4, VIM-2, NDM-1, and IMP-1). Five of these exhibited a significant inhibition of at least four enzymes, including NDM-1, VIM-2, and IMP-1. Active compounds mainly featured either halogen or bulky bicyclic aryl substituents. Finally, some compounds were also tested on several microbial dinuclear zinc-dependent hydrolases belonging to the MBL-fold superfamily (i.e., endonucleases and glyoxalase II) to explore their activity toward structurally similar but functionally distinct enzymes. Whereas the bacterial tRNases were not inhibited, the best IC50 values toward plasmodial glyoxalase II were in the 10 μm range.
Pharmacological and Biochemical Characterization of TLQP-21 Activation of a Binding Site on CHO Cells
Front Pharmacol. 2017, Mar 30;8:167. doi: 10.3389/fphar.2017.00167. eCollection 2017.
Molteni L, Rizzi L, Bresciani E, Possenti R, Petrocchi Passeri P, Ghè C, Muccioli G, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, Biagini G, Binda A, Rivolta I, Locatelli V, Torsello A.
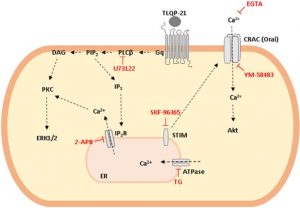
Abstract
VGF is a propeptide of 617 amino acids expressed throughout the central and the peripheral nervous system. VGF and peptides derived from its processing have been found in dense core vesicles and are released from neuronal and neuroendocrine cells via the regulated secretory pathway. Among VGF-derived neuropeptides, TLQP-21 (VGF556-576) has raised a huge interest and is one of most studied. TLQP-21 is a multifunctional neuropeptide involved in the control of several physiological functions, potentially including energy homeostasis, pain modulation, stress responsiveness and reproduction. Although little information is available about its receptor and the intracellular mechanisms mediating its biological effects, recent reports suggest that TLQP-21 may bind to the complement receptors C3aR1 and/or gC1qR. The first aim of this study was to ascertain the existence and nature of TLQP-21 binding sites in CHO cells. Secondly, we endeavored to characterize the ligand binding to these sites by using a small panel of VGF-derived peptides. And finally, we investigated the influence of TLQP-21 on selected intracellular signaling pathways. We report that CHO cells express a single class of saturable and specific binding sites for TLQP-21 with an affinity and capacity of Kd = 0.55 ± 0.05 × 10-9 M and Bmax = 81.7 ± 3.9 fmol/mg protein, respectively. Among the many bioactive products derived from the C-terminal region of VGF that we tested, TLQP-21 was the most potent in stimulating intracellular calcium mobilization in CHO cells; this effect is primarily due to its C-terminal fragment (HFHH-10). TLQP-21 induced rapid and transient dephosphorylation of phospholipase Cγ1 and phospholipase A2. Generation of IP3 and diacylglycerol was crucial for TLQP-21 bioactivity. In conclusion, our results suggest that the receptor stimulated by TLQP-21 belongs to the family of the Gq-coupled receptors, and its activation first increases membrane-lipid derived second messengers which thereby induce the mobilization of Ca2+ from the endoplasmic reticulum followed by a slower store-operated Ca2+ entry from outside the cell.
Growth hormone secretagogues prevent dysregulation of skeletal muscle calcium homeostasis in a rat model of cisplatin-induced cachexia.
J Cachexia Sarcopenia Muscle. 2017, Jun;8(3):386-404. doi: 10.1002/jcsm.12185. Epub 2017 Mar 10.
Conte E, Camerino GM, Mele A, De Bellis M, Pierno S, Rana F, Fonzino A, Caloiero R, Rizzi L, Bresciani E, Ben Haj Salah K, Fehrentz JA, Martinez J, Giustino A, Mariggiò MA, Coluccia M, Tricarico D, Lograno MD, De Luca A, Torsello A, Conte D, Liantonio A.
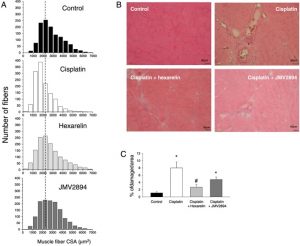
Abstract
BACKGROUND:
Cachexia is a wasting condition associated with cancer types and, at the same time, is a serious and dose-limiting side effect of cancer chemotherapy. Skeletal muscle loss is one of the main characteristics of cachexia that significantly contributes to the functional muscle impairment. Calcium-dependent signaling pathways are believed to play an important role in skeletal muscle decline observed in cachexia, but whether intracellular calcium homeostasis is affected in this situation remains uncertain. Growth hormone secretagogues (GHS), a family of synthetic agonists of ghrelin receptor (GHS-R1a), are being developed as a therapeutic option for cancer cachexia syndrome; however, the exact mechanism by which GHS interfere with skeletal muscle is not fully understood.
METHODS:
By a multidisciplinary approach ranging from cytofluorometry and electrophysiology to gene expression and histology, we characterized the calcium homeostasis in fast-twitch extensor digitorum longus (EDL) muscle of adult rats with cisplatin-induced cachexia and established the potential beneficial effects of two GHS (hexarelin and JMV2894) at this level. Additionally, in vivo measures of grip strength and of ultrasonography recordings allowed us to evaluate the functional impact of GHS therapeutic intervention.
RESULTS:
Cisplatin-treated EDL muscle fibres were characterized by a ~18% significant reduction of the muscle weight and fibre diameter together with an up-regulation of atrogin1/Murf-1 genes and a down-regulation of Pgc1-a gene, all indexes of muscle atrophy, and by a two-fold increase in resting intracellular calcium, [Ca2+ ]i , compared with control rats. Moreover, the amplitude of the calcium transient induced by caffeine or depolarizing high potassium solution as well as the store-operated calcium entry were ~50% significantly reduced in cisplatin-treated rats. Calcium homeostasis dysregulation parallels with changes of functional ex vivo (excitability and resting macroscopic conductance) and in vivo (forelimb force and muscle volume) outcomes in cachectic animals. Administration of hexarelin or JMV2894 markedly reduced the cisplatin-induced alteration of calcium homeostasis by both common as well as drug-specific mechanisms of action. This effect correlated with muscle function preservation as well as amelioration of various atrophic indexes, thus supporting the functional impact of GHS activity on calcium homeostasis.
CONCLUSIONS:
Our findings provide a direct evidence that a dysregulation of calcium homeostasis plays a key role in cisplatin-induced model of cachexia gaining insight into the etiopathogenesis of this form of muscle wasting. Furthermore, our demonstration that GHS administration efficaciously prevents cisplatin-induced calcium homeostasis alteration contributes to elucidate the mechanism of action through which GHS could potentially ameliorate chemotherapy-associated cachexia.
JMV5656, A Novel Derivative of TLQP-21, Triggers the Activation of a Calcium-Dependent Potassium Outward Current in Microglial Cells
Front Cell Neurosci. 2017 Feb 23;11:41. doi: 10.3389/fncel.2017.00041. eCollection 2017
Rivolta I, Binda A, Molteni L, Rizzi L, Bresciani E, Possenti R, Fehrentz JA, Verdié P, Martinez J, Omeljaniuk RJ, Locatelli V, Torsello A.
Abstract
TLQP-21 (TLQPPASSRRRHFHHALPPAR) is a multifunctional peptide that is involved in the control of physiological functions, including feeding, reproduction, stress responsiveness, and general homeostasis. Despite the huge interest in TLQP-21 biological activity, very little is known about its intracellular mechanisms of action. In microglial cells, TLQP-21 stimulates increases of intracellular Ca2+ that may activate functions, including proliferation, migration, phagocytosis and production of inflammatory molecules. Our aim was to investigate whether JMV5656 (RRRHFHHALPPAR), a novel short analogue of TLQP-21, stimulates intracellular Ca2+ in the N9 microglia cells, and whether this Ca2+ elevation is coupled with the activation Ca2+-sensitive K+ channels. TLQP-21 and JMV5656 induced a sharp, dose-dependent increment in intracellular calcium. In 77% of cells, JMV5656 also caused an increase in the total outward currents, which was blunted by TEA (tetraethyl ammonium chloride), a non-selective blocker of voltage-dependent and Ca2+-activated potassium (K+) channels. Moreover, the effects of ion channel blockers charybdotoxin and iberiotoxin, suggested that multiple calcium-activated K+ channel types drove the outward current stimulated by JMV5656. Additionally, inhibition of JMV5656-stimulated outward currents by NS6180 (4-[[3-(trifluoromethyl)phenyl]methyl]-2H-1,4 benzothiazin-3(4H)-one) and TRAM-34 (triarylmethane-34), indicated that KCa3.1 channels are involved in this JMV5656 mechanisms of action. In summary, we demonstrate that, in N9 microglia cells, the interaction of JMV5656 with the TLQP-21 receptors induced an increase in intracellular Ca2+, and, following extracellular Ca2+ entry, the opening of KCa3.1 channels.
Sol-gel synthesis of collagen-inspired peptide hydrogel
Materials Today , Pages: Ahead of Print, Journal, 2017, DOI: 10.1016/j.mattod.2017.02.001
C. Echalier, S. Jebors, G. Laconde, B. Guillaume, V. Luc, C. Pascal, B. Lea, A. Bethry, B. Legrand, H. Van Den Berghe, X. Garric, D. Noel, J. Martinez, A. Mehdi, G. Subra
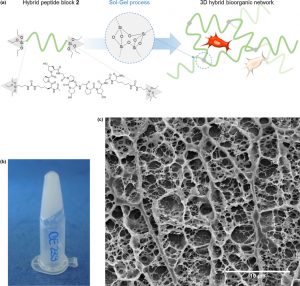
Abstract
Conceiving biomaterials able to mimic the specific environments of extracellular matrixes are a prerequisite for tissue engineering applications. Numerous types of polymers (PEG, PLA, etc.) have been used for the design of biocompatible scaffolds, but they are still less efficient than natural biopolymers such as collagen exts. Chem. modified and loaded with different bioactive factors, biopolymers afford an environment favorable to cell proliferation and differentiation. Unfortunately, they present several drawbacks, such as weak batch-to-batch reproducibility, potential immunogenicity and high cost of prodn. Herein we propose a fully synthetic covalent hydrogel obtained by sol-gel polymn. of a silylated peptide. We selected a short and low mol. building-block derived from the consensus collagen sequence [Pro-Hyp-Gly]. Interestingly, the sol-gel process occurs in physiol. buffer, enabling the embedment of stem cells. This collagen-inspired hydrogel provides a cell-friendly environment comparable to natural collagen substrates, demonstrating its potency as a biomimetic scaffold.
-Modular bioink for 3D printing of biocompatible hydrogels: sol-gel polymerization of hybrid peptides and polymers
RSC Advances, 2017, Volume: 7, Issue: 20, Pages: 12231-12235, DOI: 10.1039/C6RA28540F
C. Echalier, R. Levato, M. A. Mateos-Timoneda, O. Castano, S. Dejean, X. Garric, C. Pinese, D. Noel, E. Engel, J. Martinez, A. Mehdi, G.Subra

Abstract
An unprecedented generic system allowing the 3D printing of peptide-functionalized hydrogels by soft sol-gel inorg. polymn. is presented. Hybrid silylated inorg./bioorg. blocks are mixed in biol. buffer in an appropriate ratio, to yield a multicomponent bioink that can be printed as a hydrogel without using any photochem. or org. reagent. Hydrolysis and condensation of the silylated precursors occur during the printing process and result in a covalent network in which mols. are linked through siloxane bonds. The viscosity of the colloidal soln. used as bioink was monitored in order to set up the optimal conditions for extrusion printing. Grid-patterned hydrogel scaffolds contg. a hybrid integrin ligand were printed using a pressure-driven rapid prototyping machine. Finally, they were seeded with mesenchymal stem cells, demonstrating their suitability for cell culture. The versatility of the sol-gel process and its biocompatibility makes this approach highly promising for the prepn. of tailor-made cell-laden scaffolds.
A General Approach to the Aza-Diketomorpholine Scaffold
Organic Letters, 2017, Volume: 19, Issue: 3, Pages: 492-495, DOI: 10.1021/acs.orglett.6b03656
M. Berthet, B. Legrand, J. Martinez, I. Parrot

Abstract
A stereoconservative three-step synthesis to access to 1,2,4-oxadiazine-3,6-dione is presented. This underexplored platform could be considered as a constrained oxy-azapeptide or an aza-diketomorpholine, the methodol. being then successfully applied to produce enantiopure aza-analogs of diketomorpholine natural products. Importantly, the 1st crystal structures were obtained and compared to diketomorpholine and diketopiperazine structures. Finally, a straightforward procedure concerning the coupling of this heterocyclic scaffold with various amino acids to afford original pseudodipeptide analogs was described.
Continuous flow ring-closing metathesis, an environmentally-friendly route to 2,5-dihydro-1H-pyrrole-3-carboxylates
Green Chemistry, Pages: Ahead of Print
M. Drop, X. Bantreil, K. Grychowska, G.U. Mahoro, E. Colacino, M. Pawlowski, J. Martinez, G. Subra, P. Zajdel, F. Lamaty
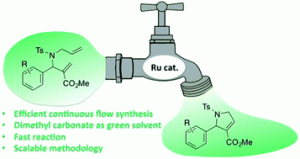
Astract
2,5-Dihydro-1H-pyrrole-3-carboxylates are important building blocks for the synthesis of high value pyrroles and pyrroloquinoline derivs. with interesting biol. activities. The use of continuous flow allowed us to perform a key synthetic step, namely ruthenium-catalyzed ring-closing metathesis, with a residence time of 1 min at 120 °C. Di-Me carbonate, a green solvent, was demonstrated for the first time to be an excellent solvent for this reaction in continuous flow. The continuous flow conditions proved to be general and the scale-up of this reaction was not only possible, but also highly efficient. Conversion of 10 g of diene was realized in 37 min under continuous flow, yielding the desired heterocycle in 91% yield.
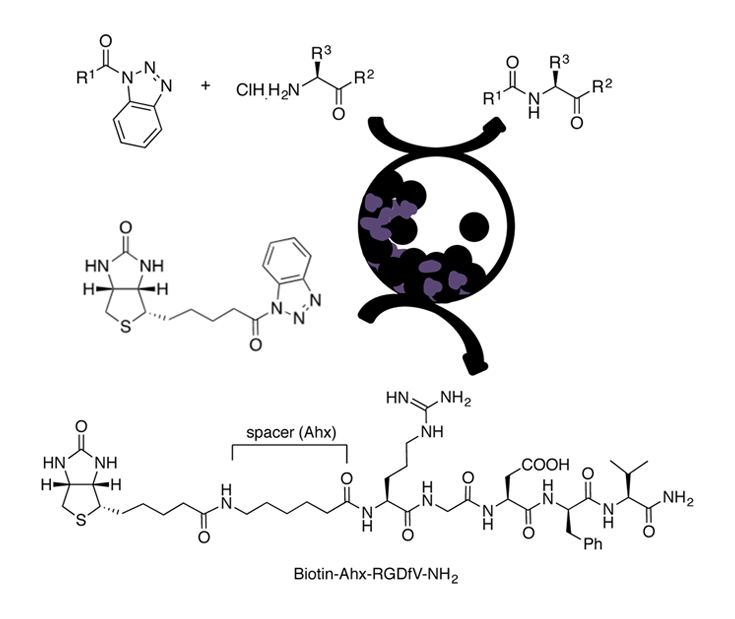
N-acyl benzotriazole derivatives for the synthesis of dipeptides and tripeptides and peptide biotinylation by mechanochemistry
ACS Sustainable Chemistry & Engineering, Pages: Ahead of Print, 2017, DOI: 10.1021/acssuschemeng.6b02439
L. Gonnet, T. Tintillier, N. Venturini, L. Konnert, J. F. Hernandez, F. Lamaty, G. Laconde, J. Martinez, E. Colacino
Abstract
An eco-friendly methodol. for prepg. Fmoc-, Z-, and Boc-N-protected dipeptides and tripeptides is described, from the corresponding N-protected-α-aminoacyl benzotriazoles and α-amino acid derivs., with different C-terminal functionalities such as esters or amides, using vibrational ball-mill (VBM). The reactivity of a β-amino ester was also investigated. In some cases, the coupling was achieved by liq.-assisted grinding (LAG). α,α- and one α,β-dipeptide were obtained in good to excellent yields mainly by pptn. in water, resulting in an improved environmental impact compared to classical peptide synthesis in soln., as shown by green metric calcns. The method was extended to the biotinylation, via an aminohexanoyl spacer, of the pentapeptide RGDfV, which contains the well-known integrins recognition site arginine-glycine-aspartic acid (RGD) motif.
JMV2894, a novel growth hormone secretagogue, accelerates body mass recovery in an experimental model of cachexia
Endocrine, 2016, Pages: Ahead of Print
E. Bresciani, L. Rizzi, L. Molteni, M. Ravelli, A. Liantonio, K. Ben Haj Salah, J. A. Fehrentz, J. Martinez, R. J. Omeljaniuk, G. Biagini, V. Locatelli, A. Torsello
Abstract
Oncol. patients subjected to chemotherapy frequently present aphagia, malnutrition, and cachexia. The purpose of this study was to investigate whether selected growth hormone secretagogues including hexarelin, JMV2894 and JMV2951 could antagonize body wt. loss and wasting induced by cisplatin administration in rats. The three growth hormone secretagogues behaved as full agonists of the growth hormone secretagogues receptor both in terms of ability to stimulate calcium mobilization in Chinese hamster ovary cells and stimulation of growth hormone release in neonatal rats. Adult rats were (i) treated with vehicle throughout (controls), or (ii) treated with cisplatin (days 1-3) and a growth hormone secretagogues or vehicle, (days 1-12). Body wt. and food consumption were measured daily. Although all growth hormone secretagogues caused initial transient acute increases in food intake, the total amt. of food eaten by controls and growth hormone secretagogues treated groups over the 12 exptl. days was not significantly different. All groups pre-treated with cisplatin lost up to 5-10 % body wt. in the first 4 days; they subsequently gained wt. at a rate comparable with controls. Interestingly, rats which received JMV2894 demonstrated a faster gain in body wt. than any other growth hormone secretagogues treated group and at the end of the protocol reached a wt. similar to that of controls. JMV2894 did not stimulate perirenal and epididymal fat accumulation but reduced MuRF mRNA levels in skeletal muscles. In conclusion, our findings demonstrate that JMV2894 antagonizes cisplatin induced wt. loss in rats and may prove useful in antagonizing cachexia assocd. with cancer and chemotherapy in humans.
Structure-Activity Relationships of JMV4463, a Vectorized Cathepsin D Inhibitor with Antiproliferative Properties: The Unique Role of the AMPA-Based Vector
ChemMedChem, 2016, Volume: 11, Issue: 3, Pages: 302-308, DOI: 10.1002/cmdc.201500457
L. Vezenkov, C. A. Sanchez, V. Bellet, V. Martin, M. Maynadier, N. Bettache, V. Lisowski, J. Martinez, M. Garcia, M. Amblard, J. F. Hernandez
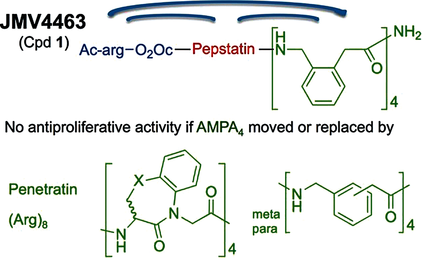
Abstract
Cathepsin D (CathD) is overexpressed and secreted by several solid tumors and stimulates their growth, the mechanism of which is still not understood. In this context, the pepstatin bioconjugate JMV4463 [Ac-arg-O2Oc-(Val)3-Sta-Ala-Sta-(AMPA)4-NH2; O2Oc=8-amino-3,6-dioxaoctanoyl, Sta=statine, AMPA=ortho-aminomethylphenylacetyl], contg. a new kind of cell-penetrating vector, was previously shown to exhibit potent antiproliferative effects in vitro and to delay the onset of tumors in vivo. In this study, the authors performed a structure-activity relationship anal. to evaluate the significance of the inhibitor and vector moieties of JMV4463. By modifying both statine residues of pepstatin the authors found that the antiproliferative activity is correlated with CathD inhibition, supporting a major role of the catalytic activity of intracellular CathD in cancer cell proliferation. Replacing the vector composed of four AMPA units with other vectors was found to abolish cytotoxicity, although all of the conjugates enabled pepstatin transport into cells. In addn., the AMPA4 vector must be localized at the C terminus of the bioconjugate. The unexpected importance of the vector structure and position for cytotoxic action suggests that AMPA4 enables pepstatin to inhibit the proteolysis of crit. CathD substrates involved in cell proliferation via a unique mechanism of action.
Unambiguous and Controlled One-Pot Synthesis of Multifunctional Silica Nanoparticles
Chemistry of Materials, 2016, Volume: 28, Issue: 3, Pages: 885-889, DOI: 10.1021/acs.chemmater.5b04398
J. Ciccione, T. Jia, J .L. Coll, K. Parra, M. Amblard, S. Jebors, J. Martinez, A. Mehdi, G. Subra
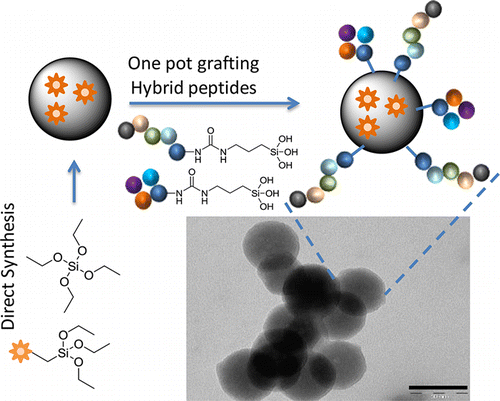
Abstract
A method for obtaining in a single step well-defined tunable multifunctional fluorescent particles having their surface functionalized with multiple covalently linked ligands is reported. The strategy relies on the synthesis of hybrid bioorg.-inorg. peptide ligands, greatly simplifying the design of multifunctional nanoparticles. It was possible to tune the ratio of two grafted ligands on the surface of the SiNPs simply by adjusting the relative concn. of hybrid species in the starting soln. An original fluorine NMR method was applied to the dissolved SiNPs to demonstrate our hypothesis.
A switchable stapled peptide
Journal of Peptide Science, 2016, Volume: 22, Issue: 3, Pages: 143-148, DOI: 10.1002/psc.2851
A. Kalistratova, B. Legrand, P. Verdie, E. Naydenova, M. Amblard, J. Martinez, G. Subra
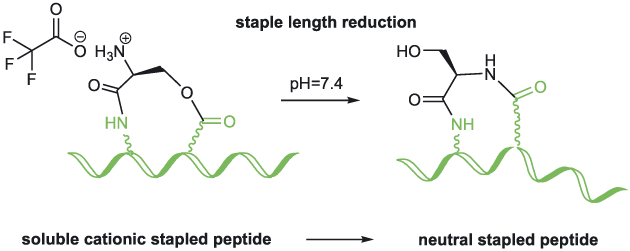
Abstract
The O-N acyl transfer reaction has gained significant popularity in peptide and medicinal chem. This reaction has been successfully applied to the synthesis of difficult sequence-contg. peptides, cyclic peptides, epimerization-free fragment coupling and more recently, to switchable peptide polymers. Herein, we describe a related strategy to facilitate the synthesis and purifn. of a hydrophobic stapled peptide. The staple consists of a serine linked through an amide bond formed from its carboxylic acid function and the side chain amino group of diaminopropionic acid and through an ester bond formed from its amino group and the side chain carboxylic acid function of aspartic acid. The α-amino group of serine was protonated during purifn. Interestingly, when the peptide was placed at physiol. pH, the free amino group initiated the O-N shift reducing the staple length by one atom, leading to a more hydrophobic stapled peptide.
Easy Synthesis of Tunable Hybrid Bioactive Hydrogels
Chemistry of Materials, 2016, Volume: 28, Issue: 5, Pages: 1261-1265, DOI: 10.1021/acs.chemmater.5b04881
C. Echalier, C. Pinese, X. Garric, H. Van Den Berghe, E. Jumas Bilak, J. Martinez, A. Mehdi, G. Subra
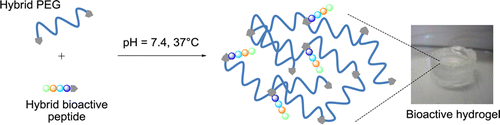
Abstract
Hydrogels are raising an increasing interest in the biomedical field and have found applications in tissue engineering and regenerative medicine. In order to mimic the complexity of natural tissues, functionalization of hydrogels with bioactive mols. is of first importance. In this context, we developed a bottom-up approach based on the synthesis of hybrid silylated blocks that can be combined to obtain covalently functionalized gels. In this study, hybrid silylated PEG and hybrid silylated bioactive peptides were synthesized and mixed in desired ratio before being simply dissolved in phosphate buff-er at physiol. pH to form a gel. The soln. turns quickly into a covalent functional gel at 37 °C. Mech. properties of these hydrogels were studied and their biocompatibility was demonstrated. Depending on the type of bioactive peptides introduced within the gels, they exhibited either antibacterial or cell adhesion properties demonstrating the potency of this sol-gel modular strategy for fine tuning of gel properties.
Selective homodimerization of unprotected peptides using hybrid hydroxydimethylsilane derivatives
RSC Advances, 2016, Volume: 6, Issue: 39, Pages: 32905-32914, DOI: 10.1039/C6RA06075G
C. Echalier, A. Kalistratova, J. Ciccione, A. Lebrun, B. Legrand, E. Naydenova, D. Gagne, J. A. Fehrentz , J. Marie, M. Amblard, A. Mehdi, J. Martinez, G. Subra
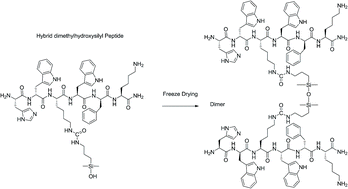
Abstract
We developed a simple and straightforward way to dimerize unprotected peptide sequences that relies on a chemoselective condensation of hybrid peptides bearing a hydroxydimethylsilyl group at a chosen position (either C-ter, N-ter or side-chain linked) to generate siloxane bonds upon freeze-drying. Interestingly, the siloxane bond sensitivity to hydrolysis is strongly pH-dependent. Thus, we investigated the stability of siloxane dimers in different exptl. conditions. For that purpose, 29Si, 13C and 1H NMR spectra were recorded to accurately quantify the ratio of dimer/monomer. More interestingly, we showed that 1H resonances of the methylene and Me groups connected to the Si can be used as sensitive probes to monitor siloxane hydrolysis and to det. the half-lives of the dimers. Importantly, we showed that the dimers were rather stable at pH 7.4 (t1/2 ≈ 400 h) and we applied the dimerization strategy to bioactive sequences. Once optimized, three dimers of the growth hormone releasing hexapeptide (GHRP-6) were prepd. Interestingly, their pharmacol. evaluation revealed that the activity of the dimeric ligands could be switched from agonist to inverse agonist depending on the position of dimerization.
New ligands of the ghrelin receptor based on the 1,2,4-triazole scaffold by introduction of a second chiral center
Bioorganic & Medicinal Chemistry Letters, 2016, Volume: 26, Issue: 10, Pages: 2408-2412, DOI: 10.1016/j.bmcl.2016.04.003
M. Maingot, A. L Blayo, S. Denoyelle, C. M’Kadmi, M. Damian, S. Mary, D. Gagne, P. Sanchez, B. Aicher, P. Schmidt, G. Muller, M. Teifel, E. Gunther, J. Marie, J.-L. Baneres, J. Martinez, J. A. Fehrentz,

Abstract
Introducing a second chiral center on the previously described 1,2,4-triazole, allowed us to increase diversity and elongate the ‘C-terminal part’ of the mol. Therefore, the authors were able to explore mimics of the substance P analogs described as inverse agonists. Some compds. presented affinities in the nanomolar range and potent biol. activities, while one exhibited a partial inverse agonist behavior similar to a Substance P analog.
Novel 1H-Pyrrolo[3,2-c]quinoline Based 5-HT6 Receptor Antagonists with Potential Application for the Treatment of Cognitive Disorders Associated with Alzheimer’s Disease
ACS Chemical Neuroscience, 2016, Volume: 7, Issue: 7, Pages: 972-983, DOI: 10.1021/acschemneuro.6b00090
K. Grychowska, G. Satala, T. Kos, A. Partyka, E. Colacino, S. Chaumont-Dubel, X. Bantreil, A. Wesolowska, M. Pawlowski, J. Martinez, P. Marin, G. Subra, A. J. Bojarski, F. Lamaty, P. Popik, P. Zajdel
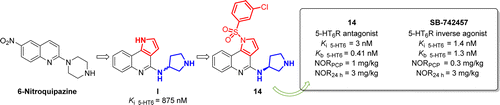
Abstract
Modulators of the serotonin 5-HT6 receptor (5-HT6R) offer a promising strategy for the treatment of the cognitive deficits that are assocd. with dementia and Alzheimer’s disease. Herein, we report the design, synthesis, and characterization of a novel class of 5-HT6R antagonists that is based on the 1H-pyrrolo[3,2-c]quinoline core. The most active compds. exhibited comparable binding affinity to the ref. compd., SB-742457, and markedly improved selectivity. Lead optimization led to the identification of (S)-1-[(3-chlorophenyl)sulfonyl]-4-(pyrrolidine-3-yl-amino)-1H-pyrrolo[3,2-c]quinoline (14) (Ki = 3 nM and Kb = 0.41 nM). Pharmacol. characterization of the 5-HT6R’s constitutive activity at Gs signaling revealed that 14 behaved as a neutral antagonist, while SB-742457 was classified as an inverse agonist. Both compds. 14 and SB-742457 reversed phencyclidine-induced memory deficits and displayed distinct procognitive properties in cognitively unimpaired animals (3 mg/kg) in NOR tasks. Compds. 14 and SB-742457 were also active in the Vogel test, yet the anxiolytic effect of 14 was 2-fold higher (MED = 3 mg/kg). Moreover, 14 produced, in a 3-fold higher dose (MED = 10 mg/kg), antidepressant-like effects that were similar to those produced by SB-742457 (MED = 3 mg/kg). Together, these data suggest that the 4-(pyrrolidine-3-yl-amino)-1H-pyrrolo[3,2-c]quinoline scaffold is an attractive mol. framework for the development of procognitive agents. The results are promising enough to warrant further detailed mechanistic studies on the therapeutic potential of 5-HT6R antagonists and inverse agonists for the treatment of cognitive decline and depression/anxiety symptoms that are comorbidities of Alzheimer’s disease.
In vivo stabilization of a gastrin-releasing peptide receptor antagonist enhances PET imaging and radionuclide therapy of prostate cancer in preclinical studies
Theranostics, 2016, Volume: 6, Issue: 1, Pages: 104-117, DOI: 10.7150/thno.13580
K. L. S. Chatalic, M. Konijnenberg, J. Nonnekens, H. de Blois, S. Erik, C. de Ridder, L. Brunel, J. A. Fehrentz, J. Martinez, D. C. van Gent, B. A. Nock, T. Maina, W. M. van Weerden, M. de Jong
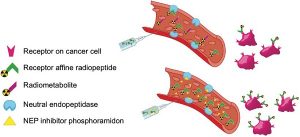
Abstract
A single tool for early detection, accurate staging, and personalized treatment of prostate cancer (PCa) would be a major breakthrough in the field of PCa. Gastrin-releasing peptide receptor (GRPR) targeting peptides are promising probes for a theranostic approach for PCa overexpressing GRPR. However, the successful application of small peptides in a theranostic approach is often hampered by their fast in vivo degrdn. by proteolytic enzymes, such as neutral endopeptidase (NEP). Here we show for the first time that co-injection of a NEP inhibitor (phosphoramidon (PA)) can lead to an impressive enhancement of diagnostic sensitivity and therapeutic efficacy of the theranostic 68Ga-/177Lu-JMV4168 GRPR-antagonist. Co-injection of PA (300 μg) led to stabilization of 177Lu-JMV4168 in murine peripheral blood. In PC-3 tumor-bearing mice, PA co-injection led to a two-fold increase in tumor uptake of 68Ga-/177Lu-JMV4168, 1 h after injection. In positron emission tomog. (PET) imaging with 68Ga-JMV4168, PA co-injection substantially enhanced PC-3 tumor signal intensity. Radionuclide therapy with 177Lu-JMV4168 resulted in significant regression of PC-3 tumor size. Radionuclide therapy efficacy was confirmed by prodn. of DNA double strand breaks, decreased cell proliferation and increased apoptosis. Increased survival rates were obsd. in mice treated with 177Lu-JMV4168 plus PA as compared to those without PA. This data shows that co-injection of the enzyme inhibitor PA greatly enhances the theranostic potential of GRPR-radioantagonists for future application in PCa patients.
12/14/14-Helix Formation in 2:1 α/β-Hybrid Peptides Containing Bicyclo[2.2.2]octane Ring Constraints
Chemistry – A European Journal, 2016, Volume: 22, Issue: 34, Pages: 11986-11990, DOI: 10.1002/chem.201602746
B. Legrand, C. Andre, L. Moulat, C. Didierjean, P. Hermet, J. L. Bantignies, J. Martinez, M. Amblard, M. Calmes
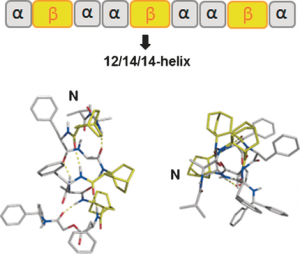
Abstract
In the search for new peptide ligands contg. selenium in their sequences, we investigated L-4-selenazolidine-carboxylic acid (selenazolidine, Sez) as a proline analog with the chalcogen atom in the γ-position of the ring. In contrast to proteinogenic selenocysteine (Sec) and selenomethionine (SeMet), the incorporation within a peptide sequence of such a non-natural amino acid has never been studied. There is thus a great interest in increasing the possibility of selenium insertion within peptides, esp. for sequences that do not possess a sulfur contg. amino acid (Cys or Met), by offering other selenated residues suitable for peptide synthesis protocols. Herein, we have evaluated selenazolidine in Boc/Bzl (Boc = tert-butoxycarbonyl) and Fmoc/tBu (Fmoc = 9-fluorenylmethoxycarbonyl) strategies through the synthesis of a model tripeptide, both in soln. and on a solid support. Special attention was paid to the stability of the Sez residue in basic conditions. Thus, generic protocols have been optimized to synthesize Sez-contg. peptides, through the use of an Fmoc-Xxx-Sez-OH dipeptide unit. As an example, a new analog of the vasopressin receptor-1A antagonist was prepd., in which Pro was replaced with Sez [3-(4-hydroxyphenyl)-propionyl-D-Tyr(Me)-Phe-Gln-Asn-Arg-Sez-Arg-NH2]. Both proline and such pseudo-proline contg. peptides exhibited similar pharmacol. properties and endopeptidase stabilities indicating that the presence of the selenium atom has minimal functional effects. Taking into account the straightforward handling of Sez as a dipeptide building block in a conventional Fmoc/tBu SPPS strategy, this result suggested a wide range of potential uses of the Sez amino acid in peptide chem., for instance as a viable proline surrogate as well as a selenium probe, complementary to Sec and SeMet, for NMR and mass spectrometry anal. purposes.
Selenazolidine: A selenium containing proline surrogate in peptide science
Organic & Biomolecular Chemistry, 2016, Volume: 14, Issue: 34, Pages: 8101-8108, DOI: 10.1039/C6OB01450J
E. Cordeau, S. Cantel, D. Gagne, A. Lebrun, J. Martinez, G. Subra, C. Enjalbal
Abstract
In the search for new peptide ligands contg. selenium in their sequences, we investigated L-4-selenazolidine-carboxylic acid (selenazolidine, Sez) as a proline analog with the chalcogen atom in the γ-position of the ring. In contrast to proteinogenic selenocysteine (Sec) and selenomethionine (SeMet), the incorporation within a peptide sequence of such a non-natural amino acid has never been studied. There is thus a great interest in increasing the possibility of selenium insertion within peptides, esp. for sequences that do not possess a sulfur contg. amino acid (Cys or Met), by offering other selenated residues suitable for peptide synthesis protocols. Herein, we have evaluated selenazolidine in Boc/Bzl (Boc = tert-butoxycarbonyl) and Fmoc/tBu (Fmoc = 9-fluorenylmethoxycarbonyl) strategies through the synthesis of a model tripeptide, both in soln. and on a solid support. Special attention was paid to the stability of the Sez residue in basic conditions. Thus, generic protocols have been optimized to synthesize Sez-contg. peptides, through the use of an Fmoc-Xxx-Sez-OH dipeptide unit. As an example, a new analog of the vasopressin receptor-1A antagonist was prepd., in which Pro was replaced with Sez [3-(4-hydroxyphenyl)-propionyl-D-Tyr(Me)-Phe-Gln-Asn-Arg-Sez-Arg-NH2]. Both proline and such pseudo-proline contg. peptides exhibited similar pharmacol. properties and endopeptidase stabilities indicating that the presence of the selenium atom has minimal functional effects. Taking into account the straightforward handling of Sez as a dipeptide building block in a conventional Fmoc/tBu SPPS strategy, this result suggested a wide range of potential uses of the Sez amino acid in peptide chem., for instance as a viable proline surrogate as well as a selenium probe, complementary to Sec and SeMet, for NMR and mass spectrometry anal. purposes.
Conformationally Constrained Peptidomimetics as Inhibitors of the Protein Arginine Methyl Transferases
Chemistry – A European Journal, 2016, Volume: 22, Issue: 39, Pages: 14022-14028, DOI: 10.1002/chem.201602518
A. Knuhtsen, B. Legrand, O. Van der Poorten, M. Amblard, J. Martinez, S. Ballet, J. L. Kristensen, D. S. Pedersen
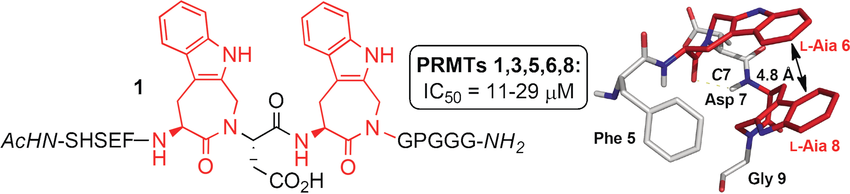
Abstract
Protein arginine N-Me transferases (PRMTs) belong to a family of enzymes that modulate the epigenetic code through modifications of histones. In the present study, peptides emerging from a phage display screening were modified in the search for PRMT inhibitors through substitution with non-proteinogenic amino acids, N-alkylation of the peptide backbone, and incorporation of constrained dipeptide mimics. One of the modified peptides (23) showed an increased inhibitory activity towards several PRMTs in the low μM range and the conformational preference of this peptide was investigated and compared with the original hit using CD and NMR spectroscopy. Introducing two constrained tryptophan residue mimics (L-Aia) spaced by a single amino acid was found to induce a unique turn structure stabilized by a hydrogen bond and arom. π-stacking interaction between the two L-Aia residues.
FT-IR and NMR structural markers for thiazole-based γ-peptide foldamers
Organic & Biomolecular Chemistry, 2016, Volume: 14, Issue: 37, Pages: 8664-8669, DOI: 10.1039/C6OB01594H
C. Bonnel, B. Legrand, J. L. Bantignies, H. Petitjean, J. Martinez, N. Masurier, L. T. Maillard
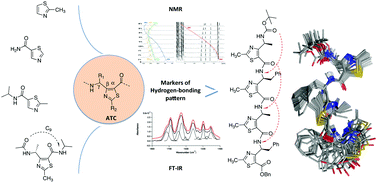
Abstract
In the search of new robust and environmental-friendly anal. methods able to answer quant. issues in pharmacol., we explore liq. chromatog. (LC) assocd. with elemental mass spectrometry (ICP-MS) to monitor peptides in such complex biol. matrixes. The novelty is to use mass spectrometry to replace radiolabelling and radioactivity measurements, which represent up-to now the gold std. to measure org. compd. concns. in life science. As a proof of concept, we choose the vasopressin (AVP)/V1A receptor system for model pharmacol. assays. The capacity of ICP-MS to provide highly sensitive quantitation of metallic and hetero elements, whatever the sample medium, prompted us to investigate this technique in combination with appropriate labeling of the peptide of interest. Selenium, that is scarcely present in biol. media, was selected as a good compromise between ICP-MS response, covalent tagging ability using conventional sulfur chem. and peptide detection specificity. Applying selenium monitoring by elemental mass spectrometry in pharmacol. is challenging due to the very high salt content and org. material complexity of the samples that produces polyat. aggregates and thus potentially mass interferences with selenium detection. Hyphenation with a chromatog. sepn. was found compulsory. Noteworthy, we aimed to develop a straightforward quant. protocol that can be performed in any lab. equipped with a std. macrobore LC-ICP-MS system, in order to avoid time-consuming sample treatment or special implementation of instrumental set-up, while allowing efficient suppression of all mass interferences to reach the targeted sensitivity. Significantly, a quantification limit of 57 ng Se L-1 (72 fmol of injected Se) was achieved, the samples issued from the pharmacol. assays being directly introduced into the LC-ICP-MS system. The established method was successfully validated and applied to the measurement of the vasopressin ligand affinity for its V1A receptor through the detn. of the dissocn. const. (Kd) which was compared to the one recorded with conventional radioactivity assays.
Investigation of elemental mass spectrometry in pharmacology for peptide quantitation at femtomolar levels
PLoS One, 2016, Volume: 11, Issue: 6, Pages: e0157943/1-e0157943/18, DOI: 10.1371/journal.pone.0157943
E. Cordeau, C. Arnaudguilhem, B. Bouyssiere, A. Hagege, J. Martinez, G. Subra, S. Cantel, C. Enjalbal

Abstract
In the search of new robust and environmental-friendly anal. methods able to answer quant. issues in pharmacol., we explore liq. chromatog. (LC) assocd. with elemental mass spectrometry (ICP-MS) to monitor peptides in such complex biol. matrixes. The novelty is to use mass spectrometry to replace radiolabelling and radioactivity measurements, which represent up-to now the gold std. to measure org. compd. concns. in life science. As a proof of concept, we choose the vasopressin (AVP)/V1A receptor system for model pharmacol. assays. The capacity of ICP-MS to provide highly sensitive quantitation of metallic and hetero elements, whatever the sample medium, prompted us to investigate this technique in combination with appropriate labeling of the peptide of interest. Selenium, that is scarcely present in biol. media, was selected as a good compromise between ICP-MS response, covalent tagging ability using conventional sulfur chem. and peptide detection specificity. Applying selenium monitoring by elemental mass spectrometry in pharmacol. is challenging due to the very high salt content and org. material complexity of the samples that produces polyat. aggregates and thus potentially mass interferences with selenium detection. Hyphenation with a chromatog. sepn. was found compulsory. Noteworthy, we aimed to develop a straightforward quant. protocol that can be performed in any lab. equipped with a std. macrobore LC-ICP-MS system, in order to avoid time-consuming sample treatment or special implementation of instrumental set-up, while allowing efficient suppression of all mass interferences to reach the targeted sensitivity. Significantly, a quantification limit of 57 ng Se L-1 (72 fmol of injected Se) was achieved, the samples issued from the pharmacol. assays being directly introduced into the LC-ICP-MS system. The established method was successfully validated and applied to the measurement of the vasopressin ligand affinity for its V1A receptor through the detn. of the dissocn. const. (Kd) which was compared to the one recorded with conventional radioactivity assays.
Application of the ring-closing metathesis to the formation of 2-aryl-1H-pyrrole-3-carboxylates as building blocks for biologically active compounds
Tetrahedron, 2016, Volume: 72, Issue: 47, Pages: 7462-7469, DOI: 10.1016/j.tet.2016.09.059
K. Grychowska, B. Kubica, M. Drop, E. Colacino, X. Bantreil, M. Pawlowski, J. Martinez, G. Subra, P. Zajdel, F. Lamaty
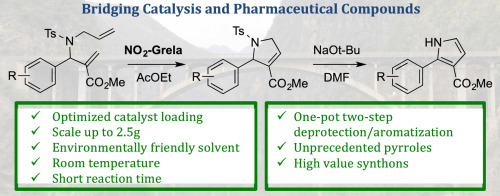
Abstract
Ring-closing metathesis (RCM) is a powerful tool for the prepn. of cyclic org. compds. Yet, one of the major limitations of this method is the difficulty to prep. large quantities of target mols. Herein we describe a comprehensive study regarding the gram-scale synthesis of 2-aryl-1H-pyrrole-3-carboxylates based on the ring-closing metathesis of the corresponding β-amino esters as a key step. This study includes evaluation of solvent and catalyst as well as reaction kinetics on the RCM. After an aromatization step, this methodol. allowed for an efficient generation of variously substituted and unprecedented 2-aryl-1H-pyrrole-3-carboxylates in good yields and cost-effectiveness. The resulting mols. might serve as key building blocks for the generation of CNS-oriented compd. libraries.
Quantitative MALDI-MS Binding Assays: An Alternative to Radiolabeling
ChemMedChem, 2016, Volume: 11, Issue: 23, Pages: 2582-2587, DOI: 10.1002/cmdc.201600447
M. Rossato, G. Miralles, C. M’Kadmi, M. Maingot, M. Amblard, B. Mouillac, D. Gagne, J. Martinez, G. Subra, C. Enjalbal, S. Cantel,
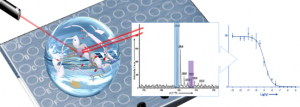
Abstract
Radiolabeling of ligands is still the gold std. in the study of high-affinity receptor-ligand interactions. In an effort toward safer and simpler alternatives to the use of radioisotopes, we developed a quant. and highly sensitive matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) method that relies on the use of chem. tagged ligands designed to be specifically detectable when present as traces in complex biol. mixts. such as cellular lysates. This innovative technol. allows easy, sensitive detection and accurate quantification of analytes at the sub-nanomolar level. After statistical validation, we were able to perform pharmacol. evaluations of G protein-coupled receptor (V1A-R)-ligand interactions. Both satn. and competitive binding assays were successfully processed.
Imidazopyridine-fused [1,3]-diazepinones part 2: Structure-activity relationships and antiproliferative activity against melanoma cells
European Journal of Medicinal Chemistry, 2016, Volume: 125, Pages: 1225-1234, DOI: 10.1016/j.ejmech.2016.11.023
V. Bellet, L. Lichon, D. P. Arama, A. Gallud, V. Lisowski, L. T. Maillard, M. Garcia, J. Martinez, N. Masurier

Abstract
We recently described a pyrido-imidazodiazepinone deriv. which could be a promising hit compd. for the development of new drugs acting against melanoma cells. In this study, a series of 28 novel pyrido-imidazodiazepinones were synthesized and screened for their in vitro cytotoxic activities against the melanoma MDA-MB-435 cell line. Among the derivs., seven of them showed 50% growth inhibitory activity at 1 μM concn., and high selectivity against the melanoma cell line MDA-MB-435.
New trisubstituted 1,2,4-triazoles as ghrelin receptor antagonists
Bioorganic & Medicinal Chemistry Letters, 2015, Volume: 25, Issue: 1, Pages: 20-24, DOI: 10.1016/j.bmcl.2014.11.031
A. Blayo, M. Maingot, B. Aicher, C. M’Kadmi, P. Schmidt, G. Muller, M. Teifel, E. Gunther, D. Gagne, S. Denoyelle, J. Martinez, J.-A. Fehrentz
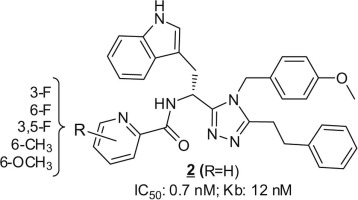
Abstract
Ghrelin receptor ligands based on a trisubstituted 1,2,4-triazole scaffold were recently synthesized and evaluated for their in vitro affinity for the GHS-R1a receptor and their biol. activity. In this study, replacement of the α-aminoisobutyryl (Aib) moiety (a common feature present in numerous growth hormone secretagogues described in the literature) by arom. and heteroarom. groups was explored. We found potent antagonists incorporating the picolinic moiety in place of the Aib moiety. In an attempt to increase affinity and activity of our lead compd. 2, we explored the modulation of the pyridine ring. Herein we report the design and the structure-activity relationships study of these new ghrelin receptor ligands.
Ghrelin receptor conformational dynamics regulate the transition from a preassembled to an active receptor:Gq complex
Proceedings of the National Academy of Sciences of the United States of America, 2015, Volume: 112, Issue: 5, Pages: 1601-1606, DOI: 10.1073/pnas.1414618112
M. Damian, S. Mary, M. Maingot, C. M’Kadmi, D. Gagne, J.-P. Leyris, S. Denoyelle, G. Gaibelet, L. Gavara, M. Costa, D. Perahia, E. Trinquet, B. Mouillac, S. Galandrin, C. Gales, J.-A. Fehrentz, N. Floquet, J. Martinez, J. Marie, J.-L. Baneres
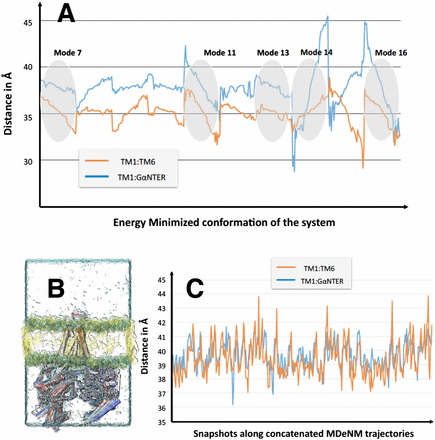
Abstract
How G protein-coupled receptor conformational dynamics control G protein coupling to trigger signaling is a key but still open question. We addressed this question with a model system composed of the purified ghrelin receptor GHS-R1a assembled into lipid disks. Combining receptor labeling through genetic incorporation of unnatural amino acids, lanthanide resonance energy transfer, and normal mode analyses, we directly demonstrate the occurrence of two distinct receptor:Gq assemblies with different geometries whose relative populations parallel the activation state of the receptor. The first of these assemblies is a preassembled complex with the receptor in its basal conformation. This complex is specific of Gq and is not obsd. with Gi. The second one is an active assembly in which the receptor in its active conformation triggers G protein activation. The active complex is present even in the absence of agonist, in a direct relationship with the high constitutive activity of the ghrelin receptor. These data provide direct evidence of a mechanism for ghrelin receptor-mediated Gq signaling in which transition of the receptor from an inactive to an active conformation is accompanied by a rearrangement of a preassembled receptor:G protein complex, ultimately leading to G protein activation and signaling.
Pyrido-imidazodiazepinones as a new class of reversible inhibitors of human kallikrein 7
European Journal of Medicinal Chemistry, 2015, Volume: 93, Pages: 202-213, DOI: 10.1016/j.ejmech.2015.02.008
D. P. Arama, F. Soualmia, V. Lisowski, J.-F. Longevial, E. Bosc, L. T. Maillard, J. Martinez, N. Masurier, C. El Amri
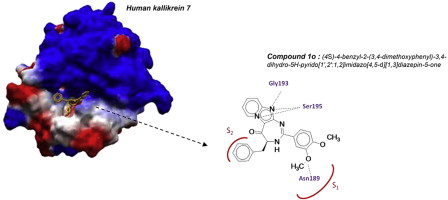
Abstract
The human tissue kallikrein-7 (KLK7) is a chymotryptic serine protease member of tissue kallikrein family. KLK7 is involved in skin homeostasis and inflammation. Excess of KLK7 activity is also assocd. with tumor metastasis processes, esp. in ovarian carcinomas, prostatic and pancreatic cancers. Development of Kallikrein 7 inhibitors is thus of great interest in oncol. but also for treating skin diseases. Most of the developed synthetic inhibitors present several drawbacks such as poor selectivity and unsuitable physico-chem. properties for in vivo use. Recently, the authors described a practical sequence for the synthesis of imidazopyridine-fused [1,3]-diazepines. Here, the authors report the identification of pyrido-imidazodiazepinone core as a new potential scaffold to develop selective and competitive inhibitors of kallikrein-related peptidase 7. Structure-activity relationships (SAR), inhibition mechanisms and selectivity as well as cytotoxicity against selected cancer cell lines were investigated.
Cross-Claisen condensation of N-Fmoc-amino acids – a short route to heterocyclic γ-amino acids
European Journal of Organic Chemistry, 2015, Volume: 2015, Issue: 10, Pages: 2262-2270, DOI: 10.1002/ejoc.201500012
L. Mathieu, C. Bonnel, N. Masurier, L. T. Maillard, J. Martinez, V. Lisowski
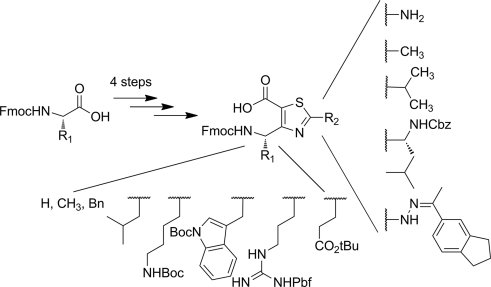
Abstract
4-Amino(methyl)-1,3-thiazole-5-carboxylic acids (ATCs) are a new class of constrained heterocyclic γ-amino acids built around a thiazole ring; these compds. are valuable as design mimics of the secondary structures of proteins such as helixes, β-sheets, turns, and β-hairpins. We report herein a short and versatile chem. route to orthogonally protected ATCs. The synthesis is centered on cross-Claisen condensations between N-Fmoc-amino acids and sterically hindered 1,1-dimethylallyl acetate. The optimized conditions are compatible with aliph., arom., acidic, and basic amino acids. The resulting N-Fmoc-β-keto ester intermediates were engaged in a two-step process to give ATCs in 45-90 % yields. The synthetic protocol provides a highly flexible method for the introduction of a wide variety of lateral chains either on the γ-carbon atom or on the thiazole core of the γ-amino acids.
A new way to silicone-based peptide polymers
Angewandte Chemie, 2015, Volume: 54, Issue: 12, Pages: 3778-3782, DOI: 10.1002/anie.201411065
S. Jebors, J. Ciccione, S. Al-Halifa, B. Nottelet, C. Enjalbal, C. M’Kadmi, M. Amblard, A. Mehdi, J. Martinez, G. Subra
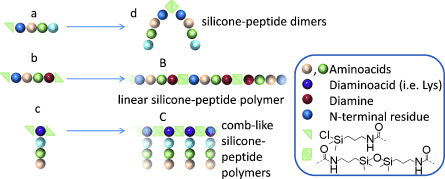
Abstract
We describe a new class of silicone-contg. peptide polymers obtained by a straightforward polymn. in water using tailored chlorodimethylsilyl peptide blocks as monomeric units. This general strategy is applicable to any type of peptide sequences, yielding linear or branched polymer chains composed of well-defined peptide sequences.
In vitro and in vivo application of radiolabeled gastrin-releasing peptide receptor ligands in breast cance
Journal of Nuclear Medicine, 2015, Volume: 56, Issue: 5, Pages: 752-757, DOI: 10.2967/jnumed.114.153023
S. U. Dalm, J. W. M. Martens, A. M. Sieuwerts, C. H. M. van Deurzen, S. J. Koelewijn, E. de Blois, T. Maina, B. A. Nock, L. Brunel, J.-A. Fehrentz, J. Martinez, M. de Jong, M. Melis
Abstract
Breast cancer (BC) consists of multiple subtypes defined by various mol. characteristics, for instance, estrogen receptor (ER) expression. Methods for visualizing BC include mammog., MR imaging, ultrasound, and nuclear medicine-based methods such as 99mTc-sestamibi and 18F-FDG PET, unfortunately all lacking specificity. Peptide receptor scintigraphy and peptide receptor radionuclide therapy are successfully applied for imaging and therapy of somatostatin receptor-expressing neuroendocrine tumors using somatostatin receptor radioligands. On the basis of a similar rationale, radioligands targeting the gastrin-releasing peptide receptor (GRP-R) might offer a specific method for imaging and therapy of BC. The aim of this study was to explore the application of GRP-R radioligands for imaging and therapy of BC by introducing valid preclin. in vitro and in vivo models. Methods: GRP-R expression of 50 clin. BC specimens and the correlation with ER expression was studied by in vitro autoradiog. with the GRP-R agonist 111In-AMBA. GRP-R expression was also analyzed in 9 BC cell lines applying 111In-AMBA internalization assays and quant. reverse transcriptase polymerase chain reaction. In vitro cytotoxicity of 111Lu-AMBA was detd. on the GRP-R-expressing BC cell line T47D. SPECT/CT imaging and biodistribution were studied in mice with s.c. and orthotopic ER-pos. T47D and MCF7 xenografts after injection of the GRP-R antagonist 111In-JMV4168. Results: Most of the human BC specimens (96%) and BC cell lines (6/9) were found to express GRP-R. GRP-R tumor expression was pos. (P = 0.026, Χ2(4) = 12,911) correlated with ER expression in the human BC specimens. Treatment of T47D cells with 10-7 M/50 MBq of 111Lu-AMBA resulted in 80% redn. of cells in vitro. Furthermore, s.c. and orthotopic tumors from both BC cell lines were successfully visualized in vivo by SPECT/CT using 111In-JMV4168; T47D tumors exhibited a higher uptake than MCF7 xenografts. Conclusion: Targeting GRP-R-expressing BC tumors using GRP-R radioligands is promising for nuclear imaging and therapy, esp. in ER-pos. BC patients.
Straightforward strategy to substitute amide bonds by 1,2,3-triazoles in peptaibols analogs using Aibψ[Tz]-Xaa dipeptides
Biopolymers, 2015, Volume: 104, Issue: 5, Pages: 611-621, DOI: 10.1002/bip.22641
K. Ben Haj Salah, B. Legrand, S. Das, J. Martinez, N. Inguimbert
Abstract
Structured peptides have gained more attention over time because of their biol. properties, biocompatibility and ability to act as modulators of protein/protein interactions, antibiotics, analgesics, immunosuppressants, and imaging agents to cite a few relevant applications. However, their poor bioavalability due in part to the susceptibility of the peptide bond to proteolytic cleavages has often impaired their development and considerably limited their therapeutic use. To circumvent these problems, many efforts have been undertaken to discover stable amide bond mimics resistant to proteolytic degrdn. Among them the 1,2,3-triazole (Tz) moiety has emerged as a highly stable analog of the trans-peptide bond as a means of generating bioactive peptides. Here we report a convenient approach to readily substitute amide bonds by triazole rings in Aib-contg. peptides using Aibψ[Tz]-Xaa dipeptide-like units. We have defined their application in solid phase synthesis and generated short model peptide sequences to study the impact of triazole incorporation on their conformations in soln. by CD and NMR spectroscopies. © 2015 Wiley Periodicals, Inc. Biopolymers (Pept Sci) 104: 611-621, 2015.
Turning Peptide Sequences into Ribbon Foldamers by a Straightforward Multicyclization Reaction
Angewandte Chemie, International Edition, 2015, Volume: 54, Issue: 47, Pages: 13966-13970, DOI: 10.1002/anie.201506955
V. Martin, B. Legrand, L. L. Vezenkov, M. Berthet, G. Subra, M. Calmes, J-L. Bantignies, J. Martinez, M. Amblard

Abstract
The conformational control of mol. scaffolds allows the display of functional groups in defined spatial arrangement. This is of considerable interest for developing fundamental and applied systems in both the fields of biol. and material sciences. Peptides afford a large diversity of functional groups, and peptide synthetic routes are very attractive and accessible. However, most short peptides do not possess well-defined secondary structures. Herein, we developed a simple strategy for converting peptide sequences into structured γ-lactam-contg. oligomers while keeping the amino acids side chain diversity. We showed the propensity of these mols. to adopt ribbon-like secondary structures. The periodic distribution of the functional groups on both sides of the ribbon plane is encoded by the initial peptide sequence.
Solid-supported synthesis and 5-HT7/5-HT1A receptor affinity of arylpiperazinylbutyl derivatives of 4,5-dihydro-1,2,4-triazine-6-(1H)-one
Chemical Biology & Drug Design, 2015, Volume: 86, Issue: 4, Pages: 697-703, DOI: 10.1111/cbdd.12539
K. Grychowska, N. Masurier, P. Verdie, G. Satala, A. J. Bojarski, J. Martinez, M. Pawlowski, G. Subra, P. Zajdel
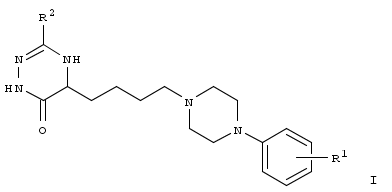
Abstract
A series of arylpiperazinylbutyl derivs. I [R1 = H, 2-OMe, 2-SMe, 2-F, 4-Cl, 2,3-diCl; R2 = Ph, 2-ClC6H4, cyclohexyl] of 4,5-dihydro-1,2,4-triazine-6(1H)-ones was designed and synthesized according to the new solid-supported methodol. In this approach, triazinone scaffold was constructed from the fmoc-protected glycine. The library representatives showed different levels of affinity for 5-HT7 and 5-HT1A receptors, among which compds. I [R1 = 2-SMe, 2-F; R2 = Ph and R1 = 2-OMe, 2-SMe, 2,3-diCl; R2 = cyclohexyl] were classified as dual 5-HT7/5-HT1A receptors ligands. The structure-affinity relationship anal. revealed that receptor affinity and selectivity of the tested compds. depended on the kind of substituent in position 3 of triazinone fragment as well as substitution pattern of phenylpiperazine moiety.
Synthesis of Thieno[3,2-e][1,4]diazepin-2-ones: Application of an Uncatalysed Pictet-Spengler Reaction
European Journal of Organic Chemistry, 2015, Volume: 2015, Issue: 32, Pages: 7146-7153, DOI: 10.1002/ejoc.201500943
S. Denoyelle, G. Tambutet, N. Masurier, L. T. Maillard, J. Martinez, V. Lisowski
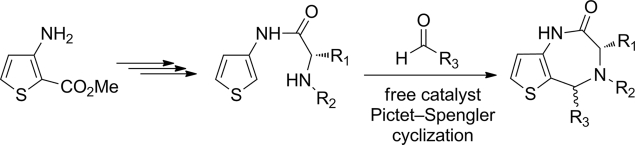
Abstract
A series of 5-substituted thieno[3,2-e][1,4]diazepin-2-ones was synthesized in four steps from Me 3-aminothiophene-2-carboxylate. After the coupling of 3-aminothiophene with α-amino acids, the key final step that involves an uncatalyzed Pictet-Spengler reaction allowed the cyclization of the seven-membered diazepinone ring. The reaction was first optimized and then exemplified in three different series (phenylalanine, alanine and proline) that led to 24 target diazepinones, which includes 19 optically pure diastereomers.
Engineered Adhesion Peptides for Improved Silicon Adsorption
Langmuir, 2015, Volume: 31, Issue: 43, Pages: 11868-11874, DOI: 10.1021/acs.langmuir.5b02857
S. R. Kumar; S. Jebors, M. Martin, T. Cloitre, V. Agarwal, A. Mehdi, J. Martinez, G. Subra, C. Gergely
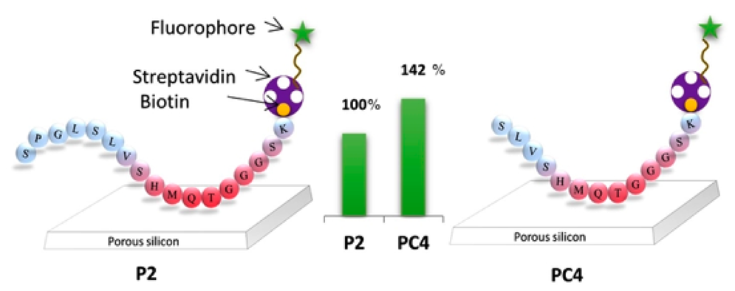
Abstract
Engineering peptides that present selective recognition and high affinity for a material is a major challenge for assembly-driven elaboration of complex systems with wide applications in the field of biomaterials, hard-tissue regeneration, and functional materials for therapeutics. Peptide-material interactions are of vital importance in natural processes but less exploited for the design of novel systems for practical applications because of our poor understanding of mechanisms underlying these interactions. Here, we present an approach based on the synthesis of several truncated peptides issued from a silicon-specific peptide recovered via phage display technol. We use the photonic response provided by porous silicon microcavities to evaluate the binding efficiency of 14 different peptide derivs. We identify and engineer a short peptide sequence (SLVSHMQT), revealing the highest affinity for p+-Si. The mol. recognition behavior of the obtained peptide fragment can be revealed through mutations allowing identification of the preferential affinity of certain amino acids toward silicon. These results constitute an advance in both the engineering of peptides that reveal recognition properties for silicon and the understanding of biomol.-material interactions.
Agonism, Antagonism, and Inverse Agonism Bias at the Ghrelin Receptor Signaling
Journal of Biological Chemistry, 2015, Volume: 290, Issue: 45, Pages: 27021-27039, DOI: 10.1074/jbc.M115.659250
C. M’Kadmi, J.-P. Leyris, L. Onfroy, C. Gales, A. Sauliere, D. Gagne, M. Damian, S. Mary, M. Maingot, S. Denoyelle, P. Verdie, J.-A. Fehrentz, J. Martinez, J.-L. Baneres, J. Marie
Abstract
The G protein-coupled receptor GHS-R1a mediates ghrelin-induced growth hormone secretion, food intake, and reward-seeking behaviors. GHS-R1a signals through Gq, Gi/o, G13, and arrestin. Biasing GHS-R1a signaling with specific ligands may lead to the development of more selective drugs to treat obesity or addiction with minimal side effects. To delineate ligand selectivity at GHS-R1a signaling, we analyzed in detail the efficacy of a panel of synthetic ligands activating the different pathways assocd. with GHS-R1a in HEK293T cells. Besides β-arrestin2 recruitment and ERK1/2 phosphorylation, we monitored activation of a large panel of G protein subtypes using a bioluminescence resonance energy transfer-based assay with G protein-activation biosensors. We first found that unlike full agonists, Gq partial agonists were unable to trigger β-arrestin2 recruitment and ERK1/2 phosphorylation. Using G protein-activation biosensors, we then demonstrated that ghrelin promoted activation of Gq, Gi1, Gi2, Gi3, Goa, Gob, and G13 but not Gs and G12. Besides, we identified some GHS-R1a ligands that preferentially activated Gq and antagonized ghrelin-mediated Gi/Go activation. Finally, we unambiguously demonstrated that in addn. to Gq, GHS-R1a also promoted constitutive activation of G13. Importantly, we identified some ligands that were selective inverse agonists toward Gq but not of G13. This demonstrates that bias at GHS-R1a signaling can occur not only with regard to agonism but also to inverse agonism. Our data, combined with other in vivo studies, may facilitate the design of drugs selectively targeting individual signaling pathways to treat only the therapeutically relevant function.
Triazole GHS-R1a antagonists JMV4208 and JMV3002 attenuate food intake, body weight, and adipose tissue mass in mice
Molecular and Cellular Endocrinology, 2014, Volume: 393, Issue: 1-2, Pages: 120-128, DOI: 10.1016/j.mce.2014.06.003
M. Holubova, V. Nagelova, Z. Lacinova, M. Haluzik, D. Sykora, A. Moulin, A. L. Blayo, J. A. Fehrentz, J. Martinez, A. Stofkova, J. Jurcovicova, B. Zelezna, L. Maletinska
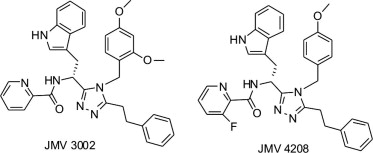
Abstract
The only peripherally released orexigenic hormone, ghrelin, plays a key role in food intake and body wt. regulation. Antagonizing the ghrelin receptor, GHS-R1a, represents a promising approach for anti-obesity therapy. In our study, two novel GHS-R1a antagonists JMV4208 and JMV3002, which are trisubstituted 1,2,4-triazoles, decreased food intake in fasted lean mice in a dose-dependent manner, with ED50 values of 5.25 and 2.05 mg/kg, resp. Both compds. were stable in mouse blood, with half-lives of 90 min (JMV4208) and 60 min (JMV3002), and disappeared from the blood 8 h after administration. Fourteen days of treatment with the ghrelin antagonists (20 mg/kg twice a day) decreased food intake, body wt. and adipose tissue mass in mice with diet-induced obesity (DIO). These results are likely attributable to an impact on food intake redn. and an attenuated expression of the lipogenesis-promoting enzymes (acetyl-CoA carboxylase 1 in s.c. fat and fatty acid synthase in s.c. and i.p. fat). The decrease in fat mass neg. impacted circulating leptin levels. These data suggest that JMV4208 and JMV3002 could be useful therapeutic agents for the treatment of obesity.
Silaproline Helical Mimetics Selectively Form an All-trans PPII Helix
Chemistry – A European Journal, 2014, Volume: 20, Issue: 44, Pages: 14240-14244, DOI: 10.1002/chem.201404820
C. Martin, B. Legrand, A. Lebrun, D. Berthomieu, J. Martinez, F. Cavelier

Abstract
The polyproline II helix (PPII) is increasingly recognized as an important element in peptide and protein structures. The discovery of pertinent PPII peptidomimetics is of great interest to tune phys. properties of the targeted structure. A series of silaproline oligomers from dimer to pentamer were synthesized. CD studies, NMR spectroscopy and mol. modeling revealed that the ribbon preferentially populates the polyproline type II secondary structure in both [D]chloroform and [D4]MeOH. The characteristics of this new lipophilic PPII-like helix were detd.
Unprecedented Chain-Length-Dependent Conformational Conversion Between 11/9 and 18/16 Helix in α/β-Hybrid Peptides
Angewandte Chemie, 2014, Volume: 53, Issue: 48, Pages: 13131-13135, DOI: 10.1002/anie.201407329
B. Legrand, C. Andre, L. Moulat, E. Wenger, C. Didierjean, E. Aubert, M. Averlant-Petit, J. Martinez, M. Calmes, M. Amblard
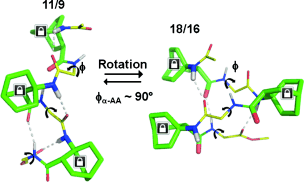
Abstract
α,β-Hybrid oligomers of varying lengths with alternating proteogenic α-amino acid and the rigid β2,3,3-trisubstituted bicyclic amino acid ABOC residues were studied using both x-ray crystal and NMR soln. structures. While only an 11/9 helix was obtained in the solid state regardless of the length of the oligomers, conformational polymorphism as a chain-length-dependent phenomenon was obsd. in soln. Consistent with DFT calcns., short oligomers adopted an 11/9 helix, whereas an 18/16 helix was favored for longer oligomers in soln. A rapid interconversion between the 11/9 helix and the 18/16 helix occurred for oligomers of intermediate length.
Preclinical comparison of Al18F- and 68Ga-labeled gastrin-releasing peptide receptor antagonists for PET imaging of prostate cancer
Journal of Nuclear Medicine, 2014, Volume: 55, Issue: 12, Pages: 2050-2056, DOI: 10.2967/jnumed.114.141143
K. l. L. S. Chatalic, G. M. Franssen, W. M. van Weerden, W. J. McBride, P. Laverman, E. de Blois, B. Hajjaj, L. Brunel, D. M. Goldenberg, J.-A. Fehrentz, J. Martinez, O. C. Boerman, M. de Jong,
Abstract
Gastrin-releasing peptide receptor (GRPR) is overexpressed in human prostate cancer and is being used as a target for mol. imaging. In this study, we report on the direct comparison of 3 novel GRPR-targeted radiolabeled tracers: Al18F-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 (JMV5132 is NODA-MPAA-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], JMV4168 is DOTA-βAla-βAla-[H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2], and NODA-MPAA is 2-[4-(carboxymethyl)-7-{[4-(carboxymethyl) phenyl]methyl}-1,4,7-triazacyclononan-1-yl]acetic acid). Methods: The GRPR antagonist JMV594 (H-D-Phe-Gln-Trp-Ala-Val-Gly-His-Sta-Leu-NH2) was conjugated to NODA-MPAA for labeling with Al18F. JMV5132 was radiolabeled with 68Ga and 18F, and JMV4168 was labeled with 68Ga for comparison. The inhibitory concn. of 50% values for binding GRPR of JMV4168, JMV5132, natGa-JMV4168, and natGa-JMV5132 were detd. in a competition-binding assay using GRPR-overexpressing PC-3 tumors. The tumor-targeting characteristics of the compds. were assessed in mice bearing s.c. PC-3 xenografts. Small-animal PET/CT images were acquired, and tracer biodistribution was detd. by ex vivo measurements. Results: JMV5132 was labeled with 18F in a novel 1-pot, 1-step procedure within 20 min, without need for further purifn. and resulting in a specific activity of 35 MBq/nmol. Inhibitory concn. of 50% values (in nM) for GRPR binding of JMV5132, JMV4168, natGa-JMV5132, natGa-JMV4168, and AlnatF-JMV5132 were 6.8 (95% confidence intervals [CIs], 4.6-10.0), 13.2 (95% CIs, 5.9-29.3), 3.0 (95% CIs, 1.5-6.0), 3.2 (95% CIs, 1.8-5.9), and 10.0 (95% CIs, 6.3-16.0), resp. In mice with s.c. PC-3 xenografts, all tracers cleared rapidly from the blood, exclusively via the kidneys for 68Ga-JMV4168 and partially hepatobiliary for 68Ga-JMV5132 and Al18F-JMV5132. Two hours after injection, the uptake of 68Ga-JMV4168, 68Ga-JMV5132, and Al18F-JMV5132 in PC-3 tumors was 5.96 ± 1.39, 5.24 ± 0.29, 5.30 ± 0.98 (percentage injected dose per g), resp. GRPR specificity was confirmed by significantly reduced tumor uptake of the 3 tracers after coinjection of a 100-fold excess of unlabeled JMV4168 or JMV5132. Small-animal PET/CT clearly visualized PC-3 tumors, with the highest resoln. obsd. for Al18F-JMV5132. Conclusion: JMV5132 could be rapidly and efficiently labeled with 18F. Al18F-JMV5132, 68Ga-JMV5132, and 68Ga-JMV4168 all showed comparable high and specific accumulation in GRPR-pos. PC-3 tumors. These new PET tracers are promising candidates for future clin. translation.
Imidazopyridine-fused [1,3]-diazepinones: Synthesis and antiproliferative activity
European Journal of Medicinal Chemistry, 2014, Volume: 75, Pages: 382-390, DOI: 10.1016/j.ejmech.2014.01.044
A. Gallud, O. Vaillant, L.T. Maillard, D. Arama, J. Dubois, M. Maynadier, V. Lisowski, M. Garcia, J. Martinez, N. Masurier
Abstract
A series of 15 pyrido-imidazo-1,3-diazepin-5-ones and pyrido-1,3-diazepine-2,5-diones were synthesized and their anticancer activities were evaluated. Among tested compds. on a cell lines panel, compd. I presents the best growth inhibition activity on 21 cell lines with a cytotoxic effect on MDA-MB-435 melanoma cells. This compd. led to deep cell morphol. changes and revealed to be an inhibitor of the Hepatocyte progenitor kinase-like kinase (HGK), which is known to be implicated in the migration, adhesion and invasion of various tumor cells.
Synthesis and reactivity of pyrrolo[3,2-d][1,3]oxazine-2,4-dione. Access to new pyrrolo[3,2-e][1,4]diazepine-2,5-diones
Tetrahedron, 2014, Volume: 70, Issue: 31, Pages: 4631-4639, DOI: 10.1016/j.tet.2014.05.046
J. Malcor, Y. Brouillette, J. Graffion, K. Spielmann, N. Masurier, L. T. Maillard, J. Martinez, V. Lisowski
Abstract
A convenient synthesis of pyrrolo[3,2-d][1,3]oxazine-2,4-dione is described and its reactivity towards various nucleophiles studied. The regioselective ring opening of pyrrolo[3,2-d][1,3]oxazine-2,4-dione or its N-alkylated analog in the presence of alanine or proline afforded, resp., imidazolidinedione and 2 N-protected pyrrolo[3,2-e][1,4]diazepines in a one-pot process. In a last part of this study, an alternative route to produce a library of eight non protected pyrrolo[3,2-e][1,4]diazepine-2,5-diones is described to overcome the limited reactivity of pyrrolo[3,2-d][1,3]oxazine-2,4-dione.
Laser desorption ionization mass spectrometry of peptides on a hybrid CHCA organic-inorganic matrix
Journal of Proteomics, Volume, 2014, 108, Pages: 369-372, DOI: 10.1016/j.jprot.2014.06.005
C. Fleith, S. Cantel, G. Subra, A. Mehdi, J. Ciccione, J. Martinez, C. Enjalbal
Abstract
We report applications of new hybrid org.-inorg. silica based materials as laser desorption/ionization (LDI)-promoting surfaces for high-throughput identification of peptides. The driving force of our work was to design a new material composed of a conventional MALDI matrix covalently attached to silica with a high org./inorg. ratio in order to improve the UV absorption by such LDI hybrid matrixes. Amorphous CHCA-functionalized silica presenting an org. content up to 1.3 mmol g-1 (around 40% in wt. from TGA and elementary anal. measurements) gave very interesting LDI performances in terms of detection sensitivity as well as relative ionization discrepancy (spectral suppression) through the analyses of small synthetic peptide mixts. (550-1300 Da) taking CHCA and amorphous silica as model matrixes for control expts.
N- and O-acetylation of threonine residues in the context of proteomics
Journal of Proteomics, Volume, 2014, 108, Pages: 369-372, DOI: 10.1016/j.jprot.2014.06.005
J.-B. Boyer, A. Dedieu, J. Armengaud, P. Verdie, G. Subra, J. Martinez, C. Enjalbal
Abstract
The detection of post-translational modifications (PTMs) of proteins is a matter of intensive research. Among all possible pitfalls that may lead to misidentifications, the chem. stability of modified peptides is scarcely questioned. Global proteomic studies devoted to protein acetylation are becoming popular. Thus, we were concerned about the intrinsic stability of O-acetylated peptides because of the O-N acyl transfer reactivity occurring when an amino moiety is present in the vicinity of the acylated hydroxyl group. Here, the behavior of isomeric O- and N-acetylated, N-terminal threonine-contg. peptides was explored in a std. proteomic workflow. We demonstrated a strong chem. instability of O-acetylation, which prevents its detection.
Ghrelin agonist JMV 1843 increases food intake, body weight and expression of orexigenic neuropeptides in mice
Physiological Research (Prague, Czech Republic), Volume: 62, Issue: 4, Pages: 435-444, ISSN: 0862-8408
M. Holubova, A. Spolcova, Z. Demianova, D. Sykora, J. A. Fehrentz, J. Martinez, A. Stofkova, J. Jurcovicova, J. Drapalova, Z. Lacinova, M. Haluzik, B. Zelezna, L. Maletinska
Abstract
Ghrelin and agonists of its receptor GHS-R1a are potential substances for the treatment of cachexia. In the present study, we investigated the acute and long-term effects of the GHS-R1a agonist JMV 1843 (H-Aib-DTrp-D-gTrp-CHO) on food intake, body wt. and metabolic parameters in lean C57BL/6 male mice. Addnl., we examd. stability of JMV 1843 in mouse blood serum. A single s.c. injection of JMV 1843 (0.01-10 mg/kg) increased food intake in fed mice in a dose-dependent manner, up to 5-times relative to the saline-treated group (ED50=1.94 mg/kg at 250 min). JMV 1843 was stable in mouse serum in vitro for 24 h, but was mostly eliminated from mouse blood after 2 h in vivo. Ten days of treatment with JMV 1843 (s.c. administration, 10 or 20 mg/kg/day) significantly increased food intake, body wt. and mRNA expression of the orexigenic neuropeptide Y and agouti-related peptide in the medial basal hypothalamus and decreased the expression of uncoupling protein 1 in brown adipose tissue. Our data suggest that JMV 1843 could have possible future uses in the treatment of cachexia.
Heating and microwave assisted SPPS of C-terminal acid peptides on trityl resin: the truth behind the yield
Amino Acids, 2013, Volume: 45, Issue: 6, Pages: 1395-1403, DOI: 10.1007/s00726-013-1604-z
C. Echalier, S. Al-Halifa, A. Kreiter, C. Enjalbal, P. Sanchez, L. Ronga, K. Puget, P. Verdie, M. Amblard, J. Martinez, G. Subra
Abstract
Despite correct purity of crude peptides prepd. on trityl resin by Fmoc/tBu microwave assisted solid phase peptide synthesis (Fmoc = 9-fluorenyl-methoxycarbonyl), surprisingly, lower yields than those expected were obtained while prepg. C-terminal acid peptides. This could be explained by cyclization/cleavage through diketopiperazine formation during the second amino acid deprotection and third amino acid coupling. However, we provide here evidence that this is not the case and that this yield loss was due to high temp. promoted hydrolysis of the 2-chloro-trityl ester, yielding premature cleavage of the C-terminal acid peptides.
Mixed Oligoureas Based on Constrained Bicyclic and Acyclic β-Amino Acids Derivatives: On the Significance of the Subunit Configuration for Folding
Source: Chemistry – A European Journal, 2013, Volume: 19, Issue: 50, Pages: 16963-16971, DOI: 10.1002/chem.201302829
C. Andre, B. Legrand, L. Moulat, E. Wenger, C. Didierjean, E. Aubert, M. Averlant-Petit, J. Martinez, M. Amblard, M. Calmes

Abstract
The combination of a nonfunctionalized constrained bicyclo[2.2.2]octane motif along with urea linkages gave a highly rigid 2.512/14 helical system both in soln. and the solid state. The authors aimed at developing stable and functionalized systems as promising materials for biol. applications in studying the impact of this constrained motif and its configuration on homo and heterochiral mixed-oligourea helix formation. Di-, tetra-, hexa-, and octa-oligoureas alternating the highly constrained bicyclic motif of (R) or (S) configuration with acyclic (S)-β3-amino acid derivs. were constructed. CD, NMR expts., and the x-ray crystal structure of the octamer unequivocally proved that the alternating heterochiral R/S sequences form a stable left-handed 2.5-helix in contrast to the mixed (S/S)-oligoureas, which did not adopt any defined secondary structure. The (-)-synclinal conformation around the Cα[n.63743]Cβ bond of the acyclic residues, although sterically less favorable than the (+)-synclinal conformation, was imposed by the (R)-bicyclic amino carbamoyl (BAC) residue. This highlighted the strong ability of the BAC residue to drive helical folding in heterochiral compds. The role of the stereochem. of the BAC unit was assessed and a model was proposed to explain the misfolding of the S/S sequences.
Thiazole-based γ-building blocks as reverse-turn mimetic to design a Gramicidin S analogue: Conformational and biological evaluation
Chemistry – A European Journal, 2013, Volume: 20, Issue: 22, Pages: 6713-6720, DOI: 10.1002/chem.201402190
B. Legrand, L. Mathieu, A. Lebrun, S. Andriamanarivo, V. Lisowski, N. Masurier, S. Zirah, Y. Kang, J. Martinez, L. Maillard

Abstract
This paper describes the ability of a new class of heterocyclic γ-amino acids named ATCs (4-amino(methyl)-1,3-thiazole-5-carboxylic acids) to induce turns when included in a tetrapeptide template. Both hybrid Ac-Val-(R or S)-ATC-Ile-Ala-NH2 sequences were synthesized and their conformations were studied by CD, NMR spectroscopy, MD simulations, and DFT calcns. It was demonstrated that the ATCs induced highly stable C9 pseudocycles in both compds. promoting a twist turn and a reverse turn conformation depending on their abs. configurations. As a proof of concept, a bioactive analog of gramicidin S was successfully designed using an ATC building block as a turn inducer. The NMR soln. structure of the analog adopted an antiparallel β-pleated sheet conformation similar to that of the natural compd. The hybrid α,γ-cyclopeptide exhibited significant reduced hemotoxicity compared to gramicidin S, while maintaining strong antibacterial activity.
Solid-supported synthesis, molecular modeling, and biological activity of long-chain arylpiperazine derivatives with cyclic amino acid amide fragments as 5-HT7 and 5-HT1A receptor ligands
European Journal of Medicinal Chemistry, 2013, Volume: 78, Pages: 10-22, DOI: 10.1016/j.ejmech.2014.03.005
V. Canale, P. Guzik, R. Kurczab, P. Verdie, G. Satala, B. Kubica, M. Pawlowski, J. Martinez, G. Subra, A. Bojarski, P. Zajdel
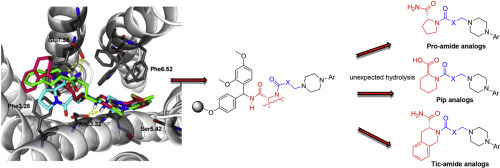
Abstract
TA 47-membered library of novel long-chain arylpiperazines, which contained cyclic amino acid amides in the terminal fragment (pyrrolidine-2-carboxamide and 1,2,3,4-tetrahydroisoquinoline-3-carboxamide), was synthesized on Rink-amide resin and biol. evaluated for binding affinity for 5-HT7 and 5-HT1A receptors. Surprisingly, members of the designed series contg. piperidine-2-carboxamide fragments underwent hydrolysis, which occurred during the acidic treatment for release from the solid-support, to their resp. pipecolic acid analogs. Representative compds. from the library displayed high-to-low affinity for 5-HT7 (Ki = 18-3134 nM) and 5-HT1A (Ki = 0.5-6307 nM) sites. The possible interactions implicated in binding of the studied compds. to the 5-HT7 receptor were supported by mol. modeling. Research was also applied to support the exploration of the influence of the amide fragment, the length of alkylene spacer, and arylpiperazine substituents on the receptor’s affinity and selectivity.
Ghrelin Receptor Ligands: Design and Synthesis of Pseudopeptides and Peptidomimetics
Current Chemical Biology, 2013, Volume: 7, Issue: 3, Pages: 254-270, , DOI: 10.2174/2212796807999131128125920
A. Moulin, L. Brunel, P. Verdie, L. Gavara, J. Martinez, J-A. Fehrentz
Abstract
A review. Mainly synthesized in the stomach, ghrelin is a peptide hormone which stimulates growth hormone secretion and appetite, thus promoting food intake and body-wt. gain. Historically, researchers started to work on the discovery of ghrelin receptor ligands several years before the discovery of the ghrelin receptor and the hormone itself. Indeed peptides able to stimulate growth hormone secretion (growth hormone releasing peptides, GHRPs) were found while the mechanism of action and the target receptor were still unknown. Non peptidic agonists were then described (growth hormone secretagogues, GHSs) and the receptor (GHS-R1a) identified in 1996. Three years later, the natural ligand of this receptor (ghrelin) was isolated from stomach and its chem. synthesis allowed to show the physiol. role of ghrelin in energy balance. In this review, we present some pseudopeptide and peptidomimetic approaches used by researchers for the design of ghrelin receptor ligands. We will start by the pioneering work of Bowers et al. on enkephalin analogs, which was the starting point for the development of an impressive no. of compds., by several of the major worldwide pharma companies. We will also describe the work achieved starting from a substance P deriv., which was one of the first peptides identified as an antagonist of the newly discovered ghrelin receptor. Then we will review the structure activity relationship study starting from the peptide ghrelin, which started with the discovery of this peptide in 1999. We will also focus on a more recent work based on macrocyclic peptidic analogs for the development of ghrelin receptor ligands.
The 1, 2, 4-triazole as a scaffold for the design of ghrelin receptor ligands: development of JMV 2959, a potent antagonist
Amino Acids. 2013 Feb;44(2):301-14. doi: 10.1007/s00726-012-1355-2. Epub 2012 Jul 14. Review.
Moulin A, Brunel L, Boeglin D, Demange L, Ryan J, M’Kadmi C, Denoyelle S, Martinez J, Fehrentz JA.
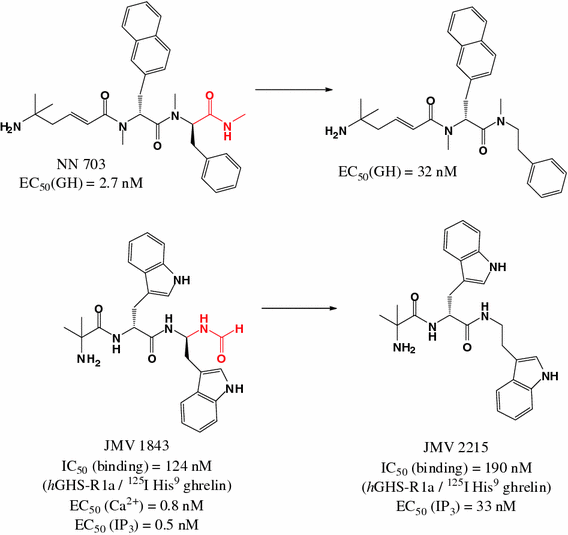
Abstract
Ghrelin is a 28-residue peptide acylated with an n-octanoyl group on the Ser 3 residue, predominantly produced by the stomach. Ghrelin displays strong growth hormone (GH) releasing activity, which is mediated by the activation of the so-called GH secretagogue receptor type 1a (GHS-R1a). Given the wide spectrum of biological activities of Ghrelin in neuroendocrine and metabolic pathways, many research groups, including our group, developed synthetic peptide, and nonpeptide GHS-R1a ligands, acting as agonists, partial agonists, antagonists, or inverse agonists. In this highlight article, we will focus on the discovery of a GHS-R1a antagonist compound, JMV 2959, which has been extensively studied in different in vitro and in vivo models. We will first describe the peptidomimetic approach that led us to discover this compound. Then we will review the results obtained with this compound in different studies in the fields of food intake and obesity, addictive behaviors, hyperactivity and retinopathy.
Dipeptide mimic oligomer transporter mediates intracellular delivery of Cathepsin D inhibitors: a potential target for cancer therapy
J Control Release. 2013 Oct 28;171(2):251-7. doi: 10.1016/j.jconrel.2013.07.017. Epub 2013 Jul 27.
Maynadier M, Vezenkov LL, Amblard M, Martin V, Gandreuil C, Vaillant O, Gary-Bobo M, Basile I, Hernandez JF, Garcia M, Martinez J.
Abstract
Implication of the intracellular proteolytic activity of Cathepsin D (CathD), a lysosomal aspartyl-protease overexpressed in numerous solid tumors, has been evidenced on tumor growth. Its intracellular inhibition by potent inhibitors such as pepstatin constitutes a relevant but challenging molecular target. Indeed the potential of pepstatin as a therapeutic molecule is hampered by its too low intracellular penetration. We addressed this limitation by designing and developing a bioconjugate combining a pepstatin derivative with a new vector of cell penetration (CPNP) specifically targeting the endolysosomal compartment. We showed that this pepstatin conjugate (JMV4463) exhibited high anti-proliferative effect on tumor cell cultures via intracellular CathD inhibition and altered cell cycle associated with apoptotic events in vitro. When tested in mice xenografted with breast cancer cells, JMV4463 delayed tumor emergence and growth.
Helical oligomers of thiazole-based γ-amino acids: synthesis and structural studies
Angew Chem Int Ed Engl. 2013 Jun 3;52(23):6006-10. doi: 10.1002/anie.201302106
Mathieu L, Legrand B, Deng C, Vezenkov L, Wenger E, Didierjean C, Amblard M, Averlant-Petit MC, Masurier N, Lisowski V, Martinez J, Maillard LT
Abstract
9-Helix: 4-Amino(methyl)-1,3-thiazole-5-carboxylic acids (ATCs) were synthesized as new γ-amino acid building blocks. The structures of various ATC oligomers were analyzed in solution by CD and NMR spectroscopy and in the solid state by X-ray crystallography. The ATC sequences adopted a well-defined 9-helix structure in the solid state and in aprotic and protic organic solvents as well as in aqueous solution.
Selective C-acylation of 2-aminoimidazo[1,2-a]pyridine: application to the synthesis of imidazopyridine-fused [1,3]diazepinones
J Org Chem. 2012 Apr 6;77(7):3679-85. doi: 10.1021/jo300364d. Epub 2012 Mar 27.
Masurier N, Aruta R, Gaumet V, Denoyelle S, Moreau E, Lisowski V, Martinez J, Maillard LT
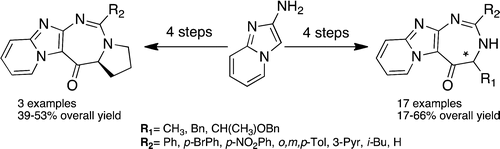
Abstract
A series of 20 optically pure 3,4-dihydro-5H-pyrido[1′,2′:1,2]imidazo[4,5-d][1,3]diazepin-5-ones which form a new family of azaheterocycle-fused [1,3]diazepines were synthesized in four steps with 17–66% overall yields. The key step consists of a selective C-acylation reaction of easily accessible 2-aminoimidazo[1,2-a]pyridine at C-3.
