
Vincent Lisowski
Professor, Faculty of Pharmacy, University OF MONTPELLIER
Vincent Lisowski was born in Aunay/Odon (France) in 1974. He first received his PharmD degree in 1998 before he obtained his PhD in 2002 from the University of Basse-Normandie, under the supervision of Professor Sylvain Rault in the field of new thiophene-based chemotherapeutic agents. After a postdoctoral position at the pharmaceutical faculty of Caen, where he developed medicinal projects in the field of cancer and Alzheimer’s disease, he joined the group of Professor Jean Martinez as an Assistant Professor at the University of Montpellier, in 2003. He was appointed Professor of Medicinal Chemistry at the School of Pharmacy in 2012. His research interests are peptide & heterocyclic chemistry applied to projects in the fields of medicinal chemistry (enzyme inhibitors, drug delivery) and chemical biology (antibody drug conjugate)
Contact:
vincent.lisowski@umontpellier.fr
0033411759599
5 major publications :
Masurier, Arama, D.P.; N; El Amri, C.; Lisowski, V. Physiological and synthetic inhibitors of tissue kallikreins: an overview. Med. Res. Rev., 2017, accepted.
Mathieu, L.; Bonnel, C.; Masurier, N.; Maillard, L. T.; Martinez, J.; Lisowski, V. Cross-Claisen Condensation of N-Fmoc-Amino Acids – A Short Route to Heterocyclic gamma-Amino Acids. European Journal of Organic Chemistry 2015, 2262-2270.
Denoyelle, S.; Tambutet, G.; Masurier, N.; Maillard, L. T.; Martinez, J.; Lisowski, V. Synthesis of Thieno[3,2-e][1,4]diazepin-2-ones: Application of an Uncatalysed Pictet-Spengler Reaction. European Journal of Organic Chemistry 2015, 7146-7153.
Legrand, B.; Mathieu, L.; Lebrun, A.; Andriamanarivo, S.; Lisowski, V.; Masurier, N.; Zirah, S.; Kang, Y. K.; Martinez, J.; Maillard, L. T. Thiazole-Based gamma-Building Blocks as Reverse-Turn Mimetic to Design a Gramicidin S Analogue: Conformational and Biological Evaluation. Chemistry-a European Journal 2014, 20, 6713-6720.
Malcor, J.-D.; Payrot, N.; David, M.; Faucon, A.; Abouzid, K.; Jacquot, G.; Floquet, N.; Debarbieux, F.; Rougon, G.; Martinez, J.; Khrestchatisky, M.; Vlieghe, P.; Lisowski, V. Chemical Optimization of New Ligands of the Low-Density Lipoprotein Receptor as Potential Vectors for Central Nervous System Targeting. Journal of Medicinal Chemistry 2012, 55, 2227-2241.

Novel thienopyrimidones targeting hepatic and erythrocytic stages of Plasmodium parasites with increased microsomal stability
Eur. J. Med. Chem., 2023, 261, 115873, https://doi.org/10.1016/j.ejmech.2023.115873
P. Lagardère, R. Mustière, N. Amanzougaghene, S. Hutter, M. Casanova, J.-F. Franetich, S. Tajeri, A. Malzert-Fréon, S. Corvaisier, M. Since, N. Azas, P. Vanelle, P. Verhaeghe, N. Primas, D. Mazier, N. Masurier, V. Lisowski
Abstract
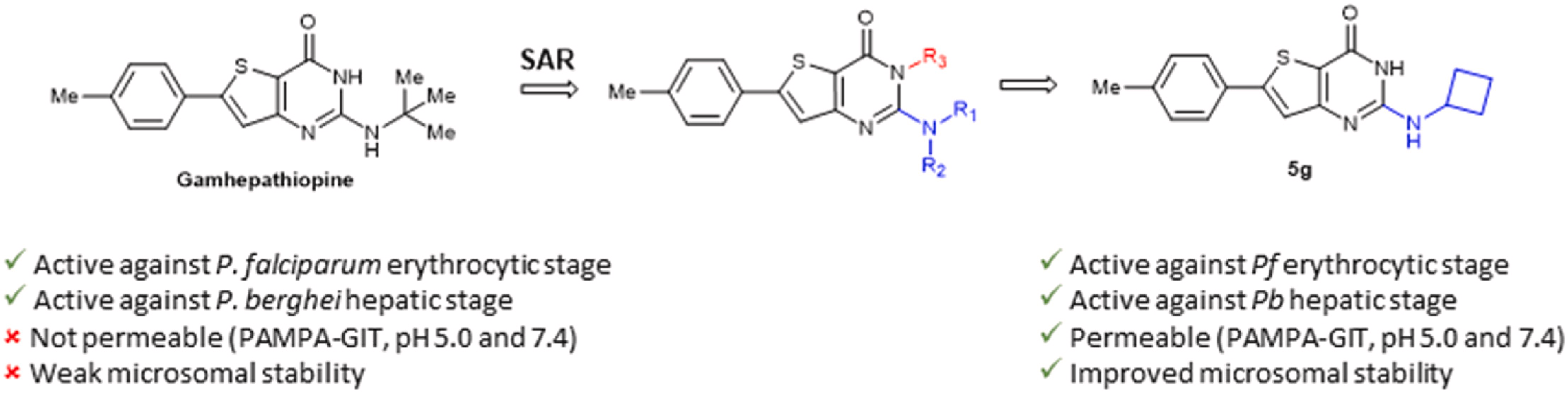
Based on the structure of a previously identified hit, Gamhepathiopine 1, which showed promising antiplasmodial activity, but poor microsomal stability, several strategies were investigated to improve the metabolic stability of the compounds. This included the introduction of fluorine or deuterium atoms, as well as carbocyclic groups. Among the new compounds, the 2-aminocyclobutyl derivative 5g demonstrated enhanced microsomal stability compared to compound 1, while retaining antiplasmodial activity against erythrocytic and hepatic stages of Plasmodium, without significant cytotoxicity against primary hepatocytes.
New antiplasmodial 4-amino-thieno[3,2-d]pyrimidines with improved intestinal permeability and microsomal stability
Eur. J. Med. Chem., 2023, 249, 115115, https://doi.org/10.1016/j.ejmech.2023.115115
P. Lagardère, R. Mustière, N. Amanzougaghene, S. Hutter, M. Casanova, J.-F. Franetich, S. Tajeri, A. Malzert-Fréon, S. Corvaisier, N. Azas, P. Vanelle, P. Verhaeghe, N. Primas, D. Mazier, N. Masurier, V. Lisowski
Abstract
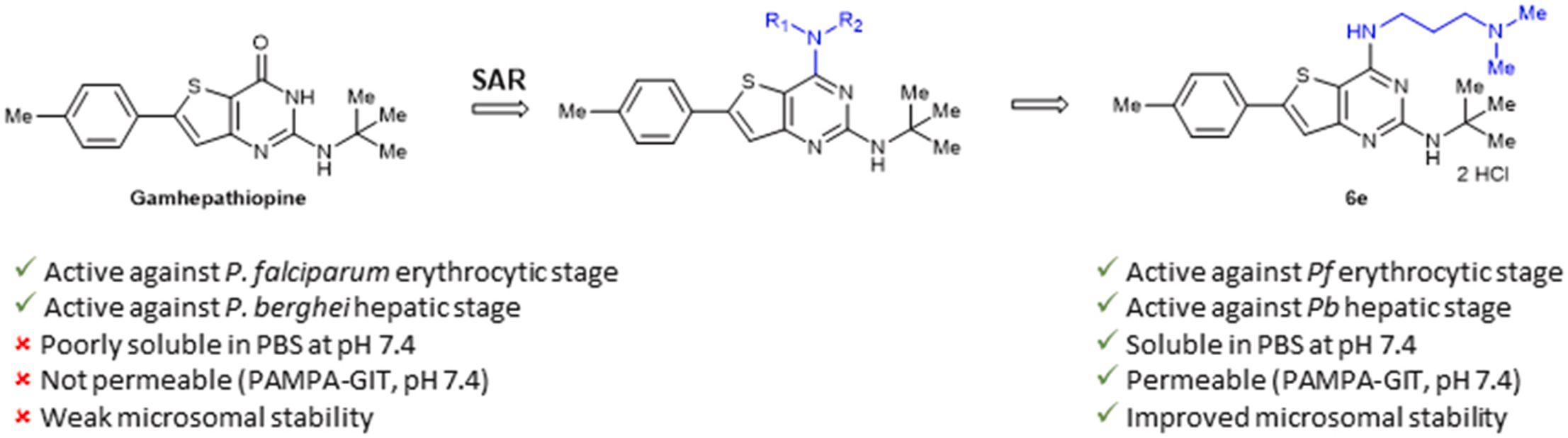
The increasing number of Plasmodium falciparum strains resistant to current treatments justifies the urgent need to discover new compounds active on several stages of the parasite development. Based on the structure of Gamhepathiopine, a 2-tert-butylaminothieno[3,2-d]pyrimidin-4(3H)-one previously identified for its dual activity against the sexual and asexual stages of P. falciparum, 25 new 4-amino-substituted analogues were synthesized and evaluated on the erythrocytic and hepatic stages of Plasmodium. A promising compound, N2-(tert-butyl)-N [4]-(3-(dimethylamino)propyl)-6-(p-tolyl)thieno[3,2-d]pyrimidine-2,4-diamine, showed improved physicochemical properties, intestinal permeability (PAMPA model) and microsomal stability compared to Gamhepathiopine, while maintaining a good antiplasmodial activity on the erythrocytic stage of P. falciparum and on the hepatic stage of P. berghei.
Synthesis of antiplasmodial 2-aminothieno[3,2-d]pyrimidin-4(3H)-one analogues using the scaffold hopping strategy
Eur. J. Med. Chem., 2022, 241, 114619, https://doi.org/10.1016/j.ejmech.2022.114619
R. Mustière, P. Lagardère, S. Hutter, V. Dell’Orco, N. Amanzougaghene, S. Tajeri, J.-F. Franetich, S. Corvaisier, Marc Since, A. Malzert-Fréon, N. Masurier, V. Lisowski, P. Verhaeghe, D. Mazier, N. Azas, P. Vanelle, N. Primas
Abstract
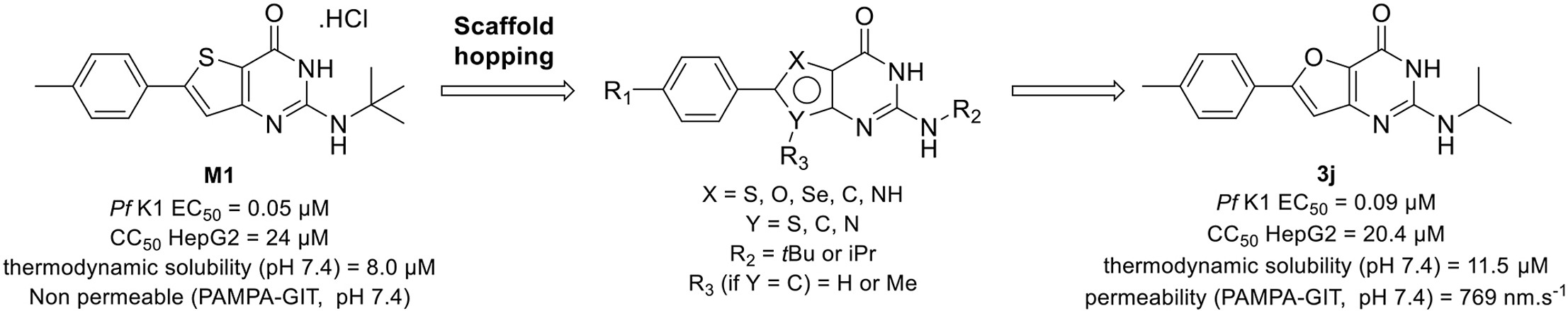
Gamhepathiopine (also known as M1), is a multi-stage acting antiplasmodial 2-tert-butylaminothieno[3,2-d]pyrimidin-4(3H)-one hydrochloride that was first described in 2015. The development of this compound is limited by poor microsomal stability, insufficient aqueous solubility and low intestinal permeability. In order to obtain new optimized derivatives, we conducted a scaffold hopping strategy from compound M1, resulting in the synthesis of 20 new compounds belonging to six chemical series. All the compounds were tested on the K1 multi-resistant strain of Plasmodium falciparum and the human HepG2 cell-line, to evaluate their antiplasmodial activity and their cytotoxicity. Analogues’ biological results also highlighted the mandatory presence of a heteroatom at position 5 of the thieno[3,2-d]pyrimidin-4(3H)-one moeity for the antiplasmodial activity. However, modifications at position 7 were detrimental for the antiplasmodial activity. We identified furane bioisostere 3j as a promising candidate, showing good blood stage antiplasmodial activity, better water solubility and highly improved intestinal permeability in the PAMPA assay.
4-Substituted Thieno[3,2-d]pyrimidines as Dual-Stage Antiplasmodial Derivatives
Pharmaceuticals, 2022, 15, 820, https://doi.org/10.3390/ph15070820
P. Lagardère, R. Mustière, N. Amanzougaghene, S. Hutter, J.-F. Franetich, N. Azas, P. Vanelle, P. Verhaeghe, N. Primas, D. Mazier, N. Masurier, V. Lisowski
Abstract
A novel series of 2-thioacetamide linked benzoxazole-benzamide conjugates 1–15 was designed as potential inhibitors of the vascular endothelial growth factor receptor-2 (VEGFR-2). The prepared compounds were evaluated for their potential antitumor activity and their corresponding selective cytotoxicity was estimated using normal human fibroblast (WI-38) cells. Compounds 1, 9–12 and 15 showed good selectivity and displayed excellent cytotoxic activity against both HCT-116 and MCF-7 cancer cell lines compared to sorafenib, used as a reference compound. Furthermore, compounds 1 and 11 showed potent VEGFR-2 inhibitory activity. The cell cycle progression assay showed that 1 and 11 induced cell cycle arrest at G2/M phase, with a concomitant increase in the pre-G1 cell population. Further pharmacological studies showed that 1 and 11 induced apoptosis and inhibited the expression of the anti-apoptotic Bcl-2 and Bcl-xL proteins in both cell lines. Therefore, compounds 1 and 11 might serve as promising candidates for future anticancer therapy development.
Thienopyrimidine: A Promising Scaffold to Access Anti-Infective Agents
Pharmaceuticals, 2022, 15(1), 35, https://doi.org/10.3390/ph15010035
P. Lagardère, C. Fersing, N. Masurier, V. Lisowski
Abstract
Thienopyrimidines are widely represented in the literature, mainly due to their structural relationship with purine base such as adenine and guanine. This current review presents three isomers—thieno[2,3-d]pyrimidines, thieno[3,2-d]pyrimidines and thieno[3,4-d]pyrimidines—and their anti-infective properties. Broad-spectrum thienopyrimidines with biological properties such as antibacterial, antifungal, antiparasitic and antiviral inspired us to analyze and compile their structure–activity relationship (SAR) and classify their synthetic pathways. This review explains the main access route to synthesize thienopyrimidines from thiophene derivatives or from pyrimidine analogs. In addition, SAR study and promising anti-infective activity of these scaffolds are summarized in figures and explanatory diagrams. Ligand–receptor interactions were modeled when the biological target was identified and the crystal structure was solved.
Pd-catalyzed C–C and C–N cross-coupling reactions in 2-aminothieno[3,2-d]pyrimidin-4(3H)-one series for antiplasmodial pharmacomodulation
RSC Advances, 2022, 12, 20004, https://doi.org/10.1039/D2RA01687G
R. Mustière, P. Lagardère, S. Hutter, C. Deraeve, F. Schwalen, D. Amrane, N. Masurier, N. Azas, V. Lisowski, P. Verhaeghe, D. Mazier, P. Vanelle, N. Primas

Abstract
In 2015, we identified gamhepathiopine (M1), a 2-tert-butylaminothieno[3,2-d]pyrimidin-4(3H)-one antiplasmodial hit targeting all development stages of the human malarial parasite P. falciparum. However, this hit compound suffers from sensitivity to hepatic oxidative metabolism. Herein, we describe the synthesis of 33 new compounds in the 2-aminothieno[3,2-d]pyrimidin-4(3H)-one series modulated at position 6 of this scaffold. The modulations were performed using three palladium-catalyzed cross coupling reactions, namely Suzuki–Miyaura, Sonogashira, and Buchwald–Hartwig. For the latter, we developed the reaction conditions. Then, we evaluated the synthesized compounds for their antiplasmodial activity on the K1 P. falciparum strain and their cytotoxicity on the human HepG2 cell line. Although we did not obtain a compound better than M1 in terms of the antiplasmodial activity, we identified compound 1g bearing a piperidine at position 6 of the thieno[3,2-d]pyrimidin-4(3H)-one ring with an improved cytotoxicity and metabolic stability. 1g is an interesting new starting point for further pharmacomodulation studies. This study also provides valuable antiplasmodial SAR data regarding the nature of the ring at position 6, the possible substituent on this ring, and the introduction of a spacer between this ring and the thienopyrimidinone moiety.
A New Thienopyrimidinone Chemotype Shows Multistage Activity against Plasmodium falciparum, Including Artemisinin-Resistant Parasites
Microbiol. Spectrum, 2021, 9 (2), e00274-21, https://journals.asm.org/doi/10.1128/spectrum.00274-21
H. Bosson-Vanga, N. Primas, J.-F. Franetich, C. Lavazec, L. Gomez, K. Ashraf, M. Tefit, V. Soulard, N. Dereuddre-Bosquet, R. Le Grand, M. Donnette, R. Mustière, N. Amanzougaghene, S. Tajeri, P. Suzanne, A. Malzert-Fréon, S. Rault, P. Vanelle, S. Hutter, A. Cohen, G. Snounou, P. Roques, N. Azas, P. Lagardère, V. Lisowski, N. Masurier, M. Nguyen, L. Paloque, F. Benoit-Vical, P. Verhaeghe, D. Mazier
Abstract
Human malaria infection begins with a one-time asymptomatic liver stage followed by a cyclic symptomatic blood stage. For decades, the research for novel antimalarials focused on the high-throughput screening of molecules that only targeted the asexual blood stages. In a search for new effective compounds presenting a triple action against erythrocytic and liver stages in addition to the ability to block the transmission of the disease via the mosquito vector, 2-amino-thienopyrimidinone derivatives were synthesized and tested for their antimalarial activity. One molecule, named gamhepathiopine (denoted as “M1” herein), was active at submicromolar concentrations against both erythrocytic (50% effective concentration [EC50] = 0.045 μM) and liver (EC50 = 0.45 μM) forms of Plasmodium falciparum. Furthermore, gamhepathiopine efficiently blocked the development of the sporogonic cycle in the mosquito vector by inhibiting the exflagellation step. Moreover, M1 was active against artemisinin-resistant forms (EC50 = 0.227 μM), especially at the quiescent stage. Nevertheless, in mice, M1 showed modest activity due to its rapid metabolization by P450 cytochromes into inactive derivatives, calling for the development of new parent compounds with improved metabolic stability and longer half-lives. These results highlight the thienopyrimidinone scaffold as a novel antiplasmodial chemotype of great interest to search for new drug candidates displaying multistage activity and an original mechanism of action with the potential to be used in combination therapies for malaria elimination in the context of artemisinin resistance.
Identification of Quinazolinone Analogs Targeting CDK5 Kinase Activity and Glioblastoma Cell Proliferation
Front Chem. 2020; 8: 691. https://doi.org/10.3389/fchem.2020.00691
Marion Peyressatre, Dominique Patomo Arama, Arthur Laure, Juan A. González-Vera, Morgan Pellerano, Nicolas Masurier, Vincent Lisowski, May C. Morris
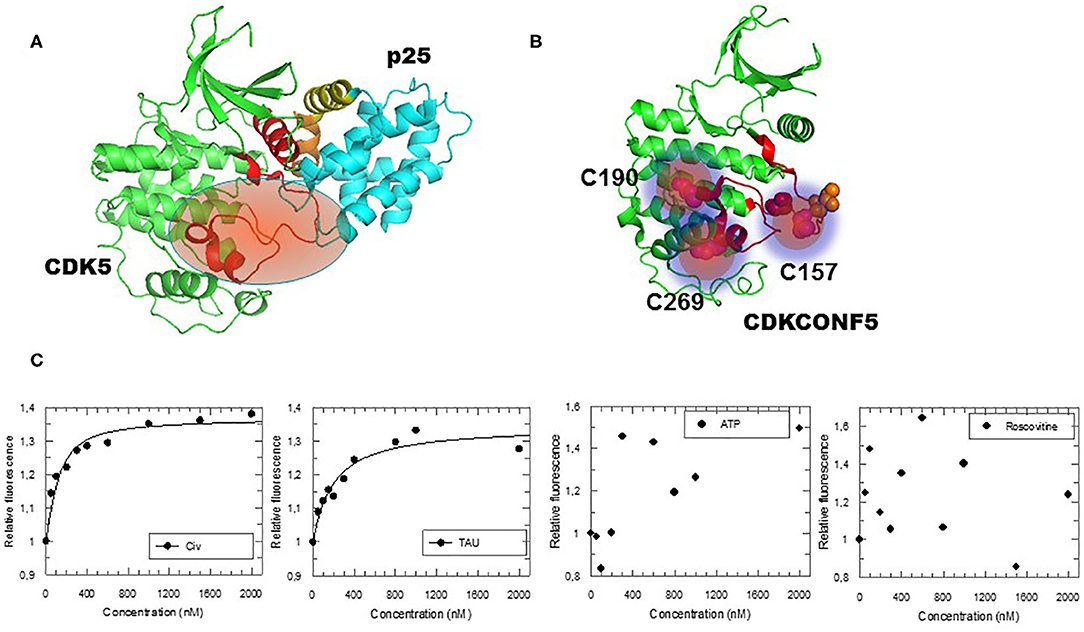
Abstract
CDK5/p25 kinase plays a major role in neuronal functions, and is hyperactivated in several human cancers including glioblastoma and neurodegenerative pathologies such as Alzheimer’s and Parkinson’s. CDK5 therefore constitutes an attractive pharmacological target. Since the successful discovery and development of Roscovitine, several ATP-competitive inhibitors of CDK5 and peptide inhibitors of CDK5/p25 interface have been developed. However, these compounds suffer limitations associated with their mechanism of action and nature, thereby calling for alternative targeting strategies. To date, few allosteric inhibitors have been developed for successful targeting of protein kinases. Indeed, although this latter class of inhibitors are believed to be more selective than compounds targeting the active site, they have proven extremely difficult to identify in high throughput screens. By implementing a fluorescent biosensor that discriminates against ATP-pocket binding compounds to screen for allosteric inhibitors that target conformational activation of CDK5, we have identified a novel family of quinazolinones. Characterization of these hits and several of their derivatives revealed their inhibitory potential toward CDK5 kinase activity in vitro and to inhibit glioblastoma cell proliferation. The quinazolinone derivatives described in this study are the first small molecules reported to target CDK5 at a site other than the ATP pocket, thereby constituting attractive leads for glioblastoma therapeutics and providing therapeutic perspectives for neurodegenerative diseases. These compounds offer alternatives to conventional ATP-competitive inhibitors or peptides targeting CDK5/p25 interface with the potential of bypassing their limitations.
The HslV Protease from Leishmania major and Its Activation by C-terminal HslU Peptides
Int J Mol Sci. 2019 Feb 26;20(5). pii: E1021. doi: 10.3390/ijms20051021
Kebe NM, Samanta K, Singh P, Lai-Kee-Him J, Apicella V, Payrot N, Lauraire N, Legrand B, Lisowski V, Mbang-Benet DE, Pages M, Bastien P, Kajava AV, Bron P, Hernandez JF, Coux O
Abstract
HslVU is an ATP-dependent proteolytic complex present in certain bacteria and in the mitochondrion of some primordial eukaryotes, including deadly parasites such as Leishmania. It is formed by the dodecameric protease HslV and the hexameric ATPase HslU, which binds via the C-terminal end of its subunits to HslV and activates it by a yet unclear allosteric mechanism. We undertook the characterization of HslV from Leishmania major (LmHslV), a trypanosomatid that expresses two isoforms for HslU, LmHslU1 and LmHslU2. Using a novel and sensitive peptide substrate, we found that LmHslV can be activated by peptides derived from the C-termini of both LmHslU1 and LmHslU2. Truncations, Ala- and D-scans of the C-terminal dodecapeptide of LmHslU2 (LmC12-U2) showed that five out of the six C-terminal residues of LmHslU2 are essential for binding to and activating HslV. Peptide cyclisation with a lactam bridge allowed shortening of the peptide without loss of potency. Finally, we found that dodecapeptides derived from HslU of other parasites and bacteria are able to activate LmHslV with similar or even higher efficiency. Importantly, using electron microscopy approaches, we observed that the activation of LmHslV was accompanied by a large conformational remodeling, which represents a yet unidentified layer of control of HslV activation.
Inhibitors of kallikrein-related peptidases: An overview.
Med Res Rev. 2018 Mar;38(2):655-683. doi: 10.1002/med.21451.
Masurier N, Arama DP, El Amri C, Lisowski V.
Abstract
Kallikrein-related peptidases (KLKs) are a family of 15 secreted serine proteases that are involved in various physiological processes. Their activities are subtly regulated by various endogenous inhibitors, ranging from metallic ions to macromolecular entities such as proteins. Furthermore, dysregulation of KLK activity has been linked to several pathologies, including cancer and skin and inflammatory diseases, explaining the numerous efforts to develop KLK-specific pharmacological inhibitors as potential therapeutic agents. In this review, we focus on the huge repertoire of KLKs inhibitors reported to date with a special emphasis on the diversity of their molecular mechanisms of inhibition.
